UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(Mark One)
THE SECURITIES EXCHANGE ACT OF 1934
OR
THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended
OR
THE SECURITIES EXCHANGE ACT OF 1934
OR
THE SECURITIES EXCHANGE ACT OF 1934
Date of event requiring this shell company report:
Commission File Number:
(Exact name of Registrant as specified in its charter)
| Not applicable | State of | |
| (Translation of Registrant’s name into English) | (Jurisdiction of incorporation or organization) |
1-800-554-9041
(Address of principal executive offices)
Robert Powell
c/o
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
| Title of each class |
Trading Symbol(s) |
Name of each exchange on which registered | ||
| The Stock Market LLC | ||||
| The Stock Market LLC |
Securities registered or to be registered pursuant to Section 12(g) of the Act: None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act: None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by this Annual Report: ordinary shares and warrants to purchase ordinary shares (as of May 1, 2024).
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| ☒ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act.
| † | The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012. |
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting over Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b).
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| US GAAP ☒ | International Financial Reporting Standards as issued by the International Accounting Standards Board ☐ | Other ☐ |
If “Other” has been checked in response to the previous question indicate by check mark which financial statement item the registrant has elected to follow. Item17 ☐ Item18 ☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐
TABLE OF CONTENTS
i
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F (including information incorporated by reference herein, the “Annual Report”) contains or may contain forward-looking statements as defined in Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that involve significant risks and uncertainties. All statements other than statements of historical facts are forward-looking statements. These forward-looking statements include information about our possible or assumed future results of operations or our performance. Words such as “expects,” “intends,” “plans,” “believes,” “anticipates,” “estimates,” and variations of such words and similar expressions are intended to identify the forward-looking statements.
Unless the context otherwise requires, all references in this Annual Report to “we,” “us”, “our” or the “Company” refer to Holdco Nuvo Group D.G Ltd. following the consummation of the Business Combination, which operates the business of Nuvo Group Ltd. and its subsidiaries following the consummation of the Business Combination, and Nuvo Group Ltd. prior to the consummation of the Business Combination.
Forward-looking statements in this Annual Report may include, for example, statements about:
| ● | the benefits of the Business Combination |
| ● | the Company’s financial performance following the Business Combination; |
| ● | the ability to maintain the listing of Holdco Ordinary Shares on the Nasdaq Global Market and the Holdco Warrants on the Nasdaq Capital Market following the Business Combination; |
| ● | the projected financial information, anticipated growth rate, and market opportunity for the Company, and estimates of expenses and profitability; |
| ● | the potential liquidity and trading of public securities of Holdco; |
| ● | the ability to raise financing in the future by Holdco; |
| ● | the effectiveness and profitability of Nuvo’s collaborations and partnerships, its ability to maintain current collaborations and partnerships and enter into new collaborations and partnerships; |
| ● | estimates related to future revenue, expenses, capital requirements and need for additional financing; |
| ● | the impact of natural disasters or health epidemics/pandemics, including a resurgence of the COVID-19 pandemic; |
| ● | the effects of increased competition as well as innovations by new and existing competitors in our industry; |
| ● | geopolitical risk, including the impacts of the ongoing conflict between Russia and Ukraine, and the war between Israel and Hamas; |
| ● | Nuvo’s ability to demonstrate the feasibility of its INVU platform for commercial applications; |
| ● | Nuvo’s ability to generate revenue in accordance with its business model; |
| ● | Nuvo’s expectations regarding its ability to obtain and maintain intellectual property protection and not infringe on the rights of others; |
| ● | Nuvo’s ability to develop, market and sell its INVU platform; |
| ● | Nuvo’s ability to develop its sales and marketing organization; |
1
| ● | changes in applicable laws or regulations; |
| ● | the outcome of any known and unknown litigation and regulatory proceedings; and |
| ● | regulatory developments in the United States and foreign countries. |
By their nature, forward-looking statements involve known and unknown risks and uncertainties because they relate to events and depend on circumstances that may or may not occur in the future and are based on potentially inaccurate assumptions. Forward-looking statements are not guarantees of future performance. The risks outlined above and others described in the section entitled (Risk Factors) are not exhaustive. Other sections of this Annual Report describe additional factors that could adversely affect the results of operations, financial condition, liquidity and the development of Nuvo and Holdco, the industry the Company operates in and risks relating to the Business Combination. New risks can emerge from time to time, and it is not possible to predict all such risks, nor can it be assessed the impact of all such risks on the Company’s business or to the extent which any such risks or combinations of risks and other factors may cause actual results to differ materially from those contained in any forward-looking statements. Given these results and uncertainties, you should not rely on forward-looking statements as a prediction of actual results.
Accordingly, you should not place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report. The Company does not undertake any obligation to publicly revise any forward-looking statement to reflect circumstances or events after the date of this Annual Report or to reflect the occurrence of unanticipated events. You should, however, review the factors and risks described in the reports filed by LAMF (prior to the SPAC Effective Time) or Holdco (after the Acquisition Effective Time) from time to time with the Securities and Exchange Commission (the “SEC”) after the date of this Annual Report.
The risk factors and cautionary language referred to or incorporated by reference in this Report provide examples of risks, uncertainties and events that may cause actual results to differ materially from the expectations described in our forward-looking statements, including among other things, the items identified in Item 3.D. “Risk Factors” of this Annual Report.
2
EXPLANATORY NOTE
On May 1, 2024, Holdco consummated the previously announced business combination pursuant to the Business Combination Agreement, dated as of August 17, 2023 (as amended, the “Business Combination Agreement”), by and among LAMF, Nuvo, Holdco, Assetco, and Merger Sub. Capitalized terms used in this section but not otherwise defined herein have the meanings given to them in the Business Combination Agreement. Pursuant to the Business Combination Agreement and the related ancillary agreements:
| ● | After the SPAC Effective Time on April 30, 2024, LAMF merged with and into Assetco (the “SPAC Merger”) with Assetco continuing as the surviving corporation (Assetco, in its capacity as the surviving entity of the SPAC Merger, the “SPAC Surviving Company”). |
| ● | Pursuant to the SPAC Merger, each Class A ordinary share of LAMF, par value $0.0001 per share (the “LAMF Class A Ordinary Shares”), issued and outstanding immediately prior to the SPAC Effective Time was automatically cancelled and converted into the right to receive outstanding ordinary shares of Holdco (“Holdco Ordinary Shares”). |
| ● | On May 1, 2024, the date of the closing of the Business Combination (the “Closing”), Merger Sub merged with and into Nuvo (the “Acquisition Merger”) with Nuvo continuing as the surviving corporation (Nuvo, in its capacity as the surviving entity of the Acquisition Merger, the “Acquisition Surviving Sub”). |
| ● | Pursuant to the Acquisition Merger, (i) each of the ordinary shares of Nuvo, par value NIS 0.01 per share (the “Nuvo Shares”), issued and outstanding immediately prior to Acquisition Effective Time were automatically cancelled and converted into the right to receive a number of Holdco Ordinary Shares determined pursuant to an equity exchange ratio of 96.139% (the “Equity Exchange Ratio”), which is equal to the equity value per share (determined by dividing an aggregate equity value of approximately $300 million upon achieving a commercial milestone (the “Equity Value”), by the fully diluted share capital of Nuvo), divided by $10.20 per share, (ii) each of the preferred shares of Nuvo, par value NIS 0.01 per share (the “Nuvo Crossover Preferred Shares”), issuable in connection with the securities purchase agreements Nuvo and Holdco entered into with certain investors prior to the execution of the Business Combination Agreement (the “Interim Financing”) issued and outstanding immediately prior to the effective time of the Acquisition Merger were automatically cancelled and converted into the right to receive a number of preferred shares of Holdco (the “Holdco Preferred Shares”) determined by the Equity Exchange Ratio, (iii) each warrant for the purchase of Nuvo Shares issued and outstanding immediately prior to the effective time of the Acquisition Merger were automatically cancelled and converted into the right to receive one warrant to purchase a number of Holdco Ordinary Shares determined by the Equity Exchange Ratio, and (iv) each outstanding and unexercised option to purchase Nuvo Shares, whether or not then vested or fully exercisable, were assumed by Holdco and converted into an option to purchase a number of Holdco Ordinary Shares as determined by the Equity Exchange Ratio, in each case subject to the adjustments described in the Business Combination Agreement. |
| ● | After the SPAC Merger and the Acquisition Merger, the SPAC Surviving Company distributed any amounts remaining in LAMF’s trust account (the “Trust Account”) to Holdco and was then liquidated (the “Liquidation”). |
The SPAC Merger, the Acquisition Merger, the Liquidation and the other transactions contemplated by the Business Combination Agreement are referred to as the “Business Combination”.
Prior to, upon and following the execution of the Business Combination Agreement, Nuvo and Holdco entered into securities purchase agreements (the “Interim Financing Agreements”) with certain investors (the “Interim Financing Investors”) pursuant to which (i) Nuvo issued Nuvo Crossover Preferred Shares to the Interim Financing Investors (which Nuvo Preferred Shares were exchanged for Holdco Preferred Shares in the Acquisition Merger) and (ii) upon the Closing, Holdco issued an aggregate of 3,823,530 Holdco Ordinary Shares to the Interim Financing Investors, which shares were not registered under the Securities Act in connection with the Business Combination Agreement, and which provided Nuvo with an aggregate of approximately $13,000,000 of gross proceeds as a result of the Interim Financing. Certain of the Interim Financing Investors are affiliated with LAMF and the Sponsor and invested an aggregate of $2,000,000 in the Interim Financing (such investors the “Sponsor Investors”). These affiliates are: (i) Jeffrey Soros, LAMF’s Chairman, who invested $500,000, (ii) Tamim Mourad, a strategic investor of LAMF and an affiliate of a member of the Sponsor, who invested $500,000 and (iii) Gaingels 10X Capital Diversity Fund I, LP, a Delaware limited partnership and an affiliate of a member of the Sponsor, that invested $1,000,000.
3
Concurrently with the consummation of the Business Combination shareholders and warrantholders of LAMF (including through units previously issued by LAMF) became shareholders and warrantholders of Holdco, other than those holders of the LAMF Class A Ordinary Shares who previously elected to redeem their LAMF Class A Ordinary Shares. The other shareholders and equityholders of Holdco include management of Nuvo and investors in Nuvo immediately before the Closing.
Bridge Financing
Since November 2023 Nuvo has been engaged in a bridge financing (the “Bridge Financing”), which involves the issuance of secured convertible bridge notes (individually, a “Bridge Financing Note”; collectively, the “Bridge Financing Notes”) to investors (“Bridge Financing Holders”).
The Bridge Financing Notes carry a 15% annual interest rate and upon conversion on the applicable Maturity Date (as defined in the Bridge Financing Notes), (i) Nuvo will pay the Holders all accrued interest on the Bridge Financing Notes up to the date of payment or conversion, and (ii) the Holders in their sole discretion, may choose to either (a) receive the principal amount of the Bridge Financing Note in cash; or (b) convert the principal amount of the investment into Nuvo Shares at a price per share of $7.0265 (or, post-Closing, the resulting number of Holdco Ordinary Shares after applying the equity exchange ratio of 96.139%).
As of the date hereof, approximately $8.5245 million in principal amount of Bridge Financing Notes has been received by Nuvo, and the offering of the Bridge Financing Notes remains ongoing.
From March 24, 2024 through April 8, 2024, Nuvo entered into amendments to all of the existing Bridge Financing Notes at that time representing $6.5732 million principal amount of the Bridge Financing Notes, to extend the maturity dates thereof (the “Bridge Financing Notes Amendments”). All new Bridge Financing Notes since April 8, 2024 include the amended maturity definition. Prior to the Bridge Financing Notes Amendments, the Bridge Financing Notes were scheduled to mature on the earlier of (i) twelve months from the issuance date thereof, (ii) the closing of the Business Combination, (iii) the closing of an initial public offering, or (iv) the closing of a bona fide financing by Nuvo for the principal purpose of raising capital, through the sale of Nuvo securities in whatever form or type (whether debt or equity) that raises in excess of $10,000,000 in gross proceeds. Pursuant to the Bridge Financing Notes Amendments, the maturity date of the amended Bridge Financing Notes was revised to be the earlier of (i) twelve months from the issuance date thereof, (ii) six (6) months following the closing of the Business Combination, (iii) six (6) months following the closing of an initial public offering, or (iv) the closing of a bona fide financing by Nuvo for the principal purpose of raising capital, through the sale of Nuvo securities in whatever form or type (whether debt or equity) that raises in excess of $25,000,000 in gross proceeds.
Each Bridge Financing Note is secured by all of Nuvo’s intellectual property, and Nuvo has filed collateral assignments/financing statements with the United States Patent & Trademark Office and is in the process of filing collateral assignments/financing statements with Nuvo’s Registrar in Israel. Gaingels 10x Capital Diversity Fund I, LP, a Bridge Financing Holder and an affiliate of a member of the Sponsor serves as collateral agent with respect to the collateral securing the Bridge Financing Notes. Upon the occurrence of any event of default described therein, the outstanding balance under the Bridge Financing Notes shall become immediately due and payable upon election of the Bridge Financing Holder and following a written demand notice sent to Nuvo.
In consideration for the services to be rendered under certain advisory services agreements between the Bridge Financing Holders and Nuvo, Nuvo issued a warrant to each Bridge Financing Holder, whereby the Bridge Financing Holder is given the right to purchase such number of Nuvo Shares (or, post-Closing, Holdco Ordinary Shares after applying the equity exchange ratio of 96.139%) equal to (2x) the principal amount of the Holder’s Bridge Financing Note divided by the same price per share noted above (i.e., $7.0265), at an exercise price of NIS 0.01.
This summary is qualified in its entirety by reference to the full text of each of the form of Bridge Financing Convertible Note, the form of Bridge Financing Warrant and the form of Bridge Financing Notes Amendment, which are filed as exhibits 4.10, 4.11 and 4.12, respectively, to this Annual Report.
Certain amounts that appear in this Annual Report may not sum due to rounding.
4
DEFINED TERMS
In this Annual Report:
“2024 Plan” means the proposed equity incentive plan for employees, directors and service providers of Holdco and its subsidiaries.
“Acquisition Effective Time” means such time as the Acquisition Merger became effective.
“Acquisition Merger” means the merger of Merger Sub with and into Nuvo.
“Amended Articles” means the amended and restated articles of association of Holdco effective immediately prior to the closing of the Business Combination.
“Assetco” means Nuvo Assetco Corp., a Cayman Islands exempted company and a wholly owned subsidiary of Holdco.
“Bridge Financing” means the bridge financing (the “Bridge Financing”) undertaken by Nuvo by issuing to investors Bridge Financing Notes since November 2023 which notes, if executed prior to April 2024, were amended in March and April 2024 to extend the maturity date thereof to no earlier than six months from the Closing.
“Bridge Financing Notes” means the secured convertible bridge notes issued in the Bridge Financing, as amended.
“Business Combination” means the Mergers and the other transactions contemplated by the Business Combination Agreement, collectively.
“Business Combination Agreement” means the Business Combination Agreement, dated as of August 17, 2023 by and among Nuvo, Holdco, Nuvo Assetco, LAMF, and Merger Sub.
“Business Day” means any day other than a Friday, a Saturday, a Sunday or other day on which commercial banks in New York, New York, Israel or the Cayman Islands are authorized or required by legal requirements to close.
“Cayman Companies Act” or “Companies Act” means Companies Act (As Revised) of the Cayman Islands.
“Closing” means the consummation of the Business Combination.
“Closing Date” means May 1, 2024, the date on which the Business Combination was consummated.
“Code” means the Internal Revenue Code of 1986, as amended.
“Companies Law” means the Israeli Companies Law, 5759-1999, as amended from time to time, including the regulations promulgated thereunder, or any other law that may come in its stead, including all amendments made thereto.
“Eligible Nuvo Equityholder” means a holder of a Nuvo Share or a Nuvo Preferred Share, in each case outstanding immediately prior to the Acquisition Effective Time.
“Equity Exchange Ratio” means the quotient obtained by dividing (a) the Equity Value Per Share by (b) the Reference Price.
“Equity Value” means an amount equal to $299,999,993.
“Equity Value Per Share” means an amount equal to (a) the Equity Value divided by (b) the number of Fully Diluted Nuvo Equity Securities.
“Exchange Act” means the Securities Exchange Act of 1934, as amended.
5
“Founder Shares” means the LAMF Class B Ordinary Shares held by Sponsor, which were converted into LAMF Class A Ordinary Shares on May 11, 2023.
“Fully Diluted Nuvo Equity Securities” means (a) the Nuvo Shares and Nuvo Preferred Shares, in each case outstanding immediately prior to the Acquisition Effective Time and (b) the Nuvo Shares that, immediately prior to the Acquisition Effective Time are issuable upon the exercise of Nuvo Warrants and Nuvo Options (whether or not vested or currently exercisable), provided, however, that Fully Diluted Nuvo Equity Securities shall not include any (i) Earnout Shares (as defined in the Business Combination Agreement) or (ii) Nuvo Shares issuable upon the conversion of then outstanding Nuvo Preferred Shares.
“Holdco” means Holdco Nuvo Group D.G Ltd., a limited liability company incorporated with limited liability under the laws of the State of Israel to serve as “Holdco” for all purposes under the Business Combination Agreement.
“Holdco Board” means the board of directors of Holdco.
“Holdco Ordinary Shares” means the ordinary shares of Holdco, no par value.
“Holdco Preferred Shares” means the preferred shares of Holdco, which shall be entitled to rights and preferences as is customary for the preferred stock of a company whose stock is traded on a national securities exchange, including those expressly set forth in the “Rights of Company Crossover Preferred Shares” attached as Exhibit E to the Business Combination Agreement and, upon conversion, they shall entitle the holder to receive Holdco Ordinary Shares.
“Holdco Securities” means collectively Holdco Ordinary Shares and Holdco Warrants.
“Holdco Shareholders” means the shareholders of Holdco.
“Holdco Warrant” means a warrant to purchase one Holdco Ordinary Share.
“IASB” means International Accounting Standards Board.
“Interim Financing” means the cross-over interim round of financing by Nuvo, whereby the Nuvo Crossover Preferred Shares were issued pursuant to the Interim Financing Agreements to the Interim Financing Investors (which Nuvo Preferred Shares were exchanged for Holdco Preferred Shares in the Acquisition Merger) and, in addition as an incentive, upon and subject to the Closing, Holdco issued Holdco Ordinary Shares to the Interim Financing Investors.
“Interim Financing Agreements” means the securities purchase agreements entered into by and between Nuvo, Holdco and the Interim Financing Investors in connection with the Interim Financing.
“Interim Financing Investors” means those certain investors in the Interim Financing.
“Investment Company Act” means the Investment Company Act of 1940, as amended.
“IPO” means LAMF’s initial public offering of LAMF Units, which was consummated on November 16, 2021.
“IRS” means the U.S. Internal Revenue Service.
“JOBS Act” means Section 2(a) of the Securities Act, as modified by the Jumpstart Our Business Startups Act of 2012, as amended.
6
“LAMF” means LAMF Global Ventures Corp. I, a Cayman Islands exempted company.
“LAMF Board” means the board of directors of LAMF.
“LAMF Class A Ordinary Shares” means LAMF’s Class A ordinary shares, par value $0.0001 per share.
“LAMF Class B Ordinary Shares” means LAMF’s Class B ordinary shares, par value $0.0001 per share.
“LAMF Exchange Ratio” means the exchange of LAMF Ordinary Shares for Holdco Ordinary Shares on a one-for-one basis.
“LAMF Insiders” means the Sponsor and certain officers and directors and advisors of LAMF.
“LAMF Ordinary Shares” means, collectively, the LAMF Class A Ordinary Shares and the LAMF Class B Ordinary Shares.
“LAMF Securities” means, collectively, the LAMF Ordinary Shares, the LAMF Warrants and the LAMF Units.
“LAMF Shareholders” means the holders of LAMF Ordinary Shares.
“LAMF Units” means the 25,300,000 LAMF units issued in connection with the IPO, each of which consists of one LAMF Class A Ordinary Share and one-half of one Public Warrant.
“LAMF Warrantholders” means holders of the LAMF Warrants.
“LAMF Warrants” means, collectively, the Public Warrants and the Private Placement Warrants.
“LAMF Warrant Agreement” means the Warrant Agreement, dated as of November 10, 2021, by and between LAMF and Continental Stock Transfer & Trust Company, as warrant agent.
“Merger Sub” means H.F.N Insight Merger Company Ltd., a limited liability company organized under the laws of the State of Israel and a wholly owned subsidiary of LAMF.
“Mergers” means the Acquisition Merger and the SPAC Merger.
“Nasdaq” means the Nasdaq Global Market.
“Nuvo” means Nuvo Group Ltd., a limited liability company organized under the laws of the State of Israel.
“Nuvo 2015 Plan” means Nuvo’s 2015 Share Incentive Plan.
“Nuvo Crossover Preferred Shares” means the preferred shares of Nuvo, with par value NIS 0.01 per share, issued in connection with the Interim Financing.
“Nuvo Convertible Loans” means the convertible loans made by certain investors pursuant to several loan agreements entered into from May 29, 2022 through June 30, 2023 (as amended in August 2023 in connection with the execution of the Business Combination Agreement), by and between Nuvo and each such investor, which loans represented an aggregate principal amount of approximately $7.9 million bear interest at a rate of 2% per month, matured on the Closing Date, at which time the principal amount and accrued interest on such loans were applied to the related Nuvo SAFEs issued to such investors in connection with provision of the Nuvo Convertible Loans.
“Nuvo Loan Amendment” means the amendments to the Nuvo Convertible Loans to cause each Nuvo Convertible Loan to be automatically converted prior to the Acquisition Effective Time into Nuvo Shares pursuant to the terms of such Nuvo Convertible Loan and under the terms of the Nuvo SAFE Amendment.
7
“Nuvo Options” means each outstanding and unexercised option to purchase Nuvo Shares, whether or not then vested or fully exercisable, granted prior to the Acquisition Effective Time to any current or former employee, officer, director or other service provider of Nuvo or its direct and indirect subsidiaries.
“Nuvo Optionholders” means the holders of the Nuvo Options.
“Nuvo Preferred Shares” means the Nuvo Crossover Preferred Shares.
“Nuvo SAFEs” means the Simple Agreements for Future Equity of the Company entered into by and between Nuvo and certain investors, service providers and lenders, from June 2020 through April 2023 (as amended in August 2023 pursuant to the Nuvo SAFE Amendment).
“Nuvo SAFE Amendment” means the amendments to cause each Nuvo SAFE to be automatically converted prior to the Acquisition Effective Time into Nuvo Shares pursuant to the terms of such Nuvo SAFEs.
“Nuvo Shares” means the ordinary shares of Nuvo, with par value NIS 0.01 per share.
“Nuvo Shareholders” means the shareholders of Nuvo.
“Nuvo Warrants” means the warrants issued on May 20, 2015 by Nuvo, exercisable to purchase up to 45,428 Nuvo Shares at an exercise price per share of NIS 0.01.
“Original Registration Rights Agreement” means that certain Registration Rights Agreement, dated as of November 10, 2021, by and among LAMF, Sponsor and certain other parties thereto.
“PFIC” means passive foreign investment company.
“Private Placement Units” means the 1,106,000 private placement units, purchased by the Sponsor at a price of $10.00 per Private Placement Unit in a private placement consummated concurrently with the closing of the IPO, each consisting of one LAMF Class A Ordinary Share and one-half of one Private Placement Warrant.
“Private Placement Warrants” means the warrants to purchase LAMF Class A Ordinary Shares purchased in a private placement in connection with the IPO, at an exercise price of $11.50 per share.
“Pro Rata Share” means, for each Eligible Nuvo Equityholder, a percentage determined by dividing (a) the sum of (i) the total number of Nuvo Shares issued and outstanding held by such Eligible Nuvo Equityholder immediately prior to the Acquisition Effective Time, plus (ii) the total number of Nuvo Preferred Shares issued and outstanding held by such Eligible Nuvo Equityholder immediately prior to the Acquisition Effective Time, by (b) the total number of Nuvo Shares and Nuvo Preferred Shares issued and outstanding as of immediately prior to the Acquisition Effective Time.
“Public Shareholders” means the holders of the Public Shares.
“Public Shares” means the LAMF Class A Ordinary Shares sold in the IPO (whether such shares were purchased in the IPO as part of the LAMF Units or thereafter in the open market).
“Public Warrants” means the warrants included in the LAMF Units sold in the IPO, each of which is exercisable for one LAMF Class A Ordinary Share, in accordance with its terms, at an exercise price of $11.50 per share.
“Redemption Right” means the right to redeem LAMF Class A Ordinary Shares in connection with the approval of the Business Combination.
“Reference Price” means $10.20.
“Registration Rights Agreement” means the registration rights agreement, dated as of May 1, 2024, by and among Holdco, Nuvo, LAMF, Sponsor, certain affiliates and members of the Sponsor and certain Nuvo Shareholders, which is in the form attached to the Business Combination Agreement as Exhibit C.
8
“SEC” means the U.S. Securities and Exchange Commission.
“Securities Act” means the Securities Act of 1933, as amended.
“Shareholder Support Agreement” means the Shareholder Support Agreement, dated as of August 17, 2023 by and among LAMF, the Nuvo Shareholders, Nuvo and Holdco.
“SPAC Effective Time” means such time as the SPAC Merger becomes effective.
“SPAC Exchange Ratio” means 1.00.
“SPAC Merger” means the merger of LAMF with and into Assetco upon the terms and subject to the conditions set forth in the Business Combination Agreement, the plan of merger relating to the SPAC Merger and in accordance with the applicable provisions of the Companies Act, whereupon the separate corporate existence of LAMF ceased and Assetco continued its existence under the Companies Act as the surviving company.
“Sponsor” means LAMF SPAC Holdings I LLC, a Cayman Islands limited liability company.
“Sponsor Shares” means the LAMF Class A Ordinary Shares and LAMF Class B Ordinary Shares held by Sponsor.
“Sponsor Support Agreement” means the Sponsor Support Agreement, dated as of August 17, 2023 by and among LAMF, Nuvo, Holdco, Sponsor and the LAMF directors and executive officers signatories thereto.
“Trading Day” means any day on which Holdco Ordinary Shares are tradeable on Nasdaq (or the principal securities exchange or securities market on which Holdco Ordinary Shares are then traded).
“Transaction Documents” means, collectively, the Business Combination Agreement, the Sponsor Support Agreement, the Shareholder Support Agreement, Registration Rights Agreement, the Amended Articles, the Interim Financing Agreements, the Warrant Assignment, Assumption and Amendment Agreement and all the agreements, documents, instruments and certificates entered into in connection therewith and any and all exhibits and schedules thereto.
“Transaction Expenses” means to the extent not paid prior to Closing, all out-of-pocket fees, costs and expenses of counsel, accountants, investment bankers, experts and consultants to a party to the Business Combination Agreement incurred by such party or on its behalf in connection with the consummation of the Transactions or related to the authorization, preparation, negotiation, execution and performance of the Business Combination Agreement.
“Transactions” means, collectively, the Mergers and each of the other transactions contemplated by the Business Combination Agreement or any of the other Transaction Documents.
“Trust Account” means the U.S.-based trust account at J.P. Morgan Chase Bank, N.A., with Continental acting as trustee, that held a portion of the proceeds of the IPO and the concurrent sale of the Private Placement Warrants.
“U.S.” means the United States.
“U.S. GAAP” means generally accepted accounting principles in the United States as are in effect from time to time.
“Warrant Assignment, Assumption and Amendment Agreement” means the warrant assignment, assumption and amendment agreement entered into by and among LAMF, Holdco and Continental at the SPAC Effective Time, pursuant to which LAMF assigned all its rights, title and interest in the LAMF Warrant Agreement to Holdco.
“Working Capital Loans” mean the $550,000 principal amount outstanding as of the Closing under the unsecured convertible promissory note issued by LAMF to the Sponsor on February 2, 2024, which converted pursuant to the terms of such note into 55,000 private placement units of LAMF, consisting of 55,000 LAMF Class A Ordinary Shares and 27,500 private LAMF Warrants immediately prior to the Closing.
9
RISK FACTOR SUMMARY
Investing in our securities entails a high degree of risk as more fully described under “Risk Factors.” You should carefully consider such risks before deciding to invest in our securities. These risks include, among others:
| ● | Nuvo is a development-stage company with a limited operating history, and may never be able to effectuate its business plan, achieve meaningful revenue or attain profitability. |
| ● | Nuvo is highly dependent on the successful development, marketing and sale of the INVU platform and the related products and services. |
| ● | Nuvo will need to obtain additional financing to fund its future operations and continue as a going concern. |
| ● | The manufacturing and supply of the INVU platform is subject to various factors outside Nuvo’s direct control, including those related to Nuvo’s dependence on third-party manufacturers and suppliers. |
| ● | Nuvo’s medical device operations are subject to pervasive and continuing FDA regulatory requirements, and failure to comply with these requirements could harm its business, financial condition and results of operations. |
| ● | Healthcare reform initiatives and other administrative and legislative proposals may harm Nuvo’s business. |
| ● | The results of Nuvo’s clinical trials may not support the INVU platform claims or may result in the discovery of adverse side effects. |
| ● | Conditions in Israel could materially and adversely affect Nuvo’s business. |
| ● | Nuvo may be unable to obtain and maintain patent or other intellectual property protection for any product it develops for its technology. |
| ● | Holdco will incur increased costs as a result of operating as a public company. |
| ● | A market for Holdco Ordinary Shares may not develop, which would adversely affect the liquidity and price of Holdco Ordinary Shares. |
| ● | The price of Holdco Ordinary Shares may be volatile. |
| ● | It is not expected that Holdco will pay dividends in the foreseeable future after the Business Combination. |
| ● | Holdco may not be able to timely and effectively implement controls and procedures required by Section 404(a) of the Sarbanes-Oxley Act that will be applicable to it. |
| ● | As a foreign private issuer and a company treated as an emerging growth company for certain purposes, Holdco has different disclosure and other requirements than U.S. domestic registrants and non-emerging growth companies. |
| ● | Holdco may lose its foreign private issuer status, which would then require Holdco to comply with the Exchange Act’s domestic reporting regime and cause Holdco to incur significant legal, accounting and other expenses. |
| ● | If securities or industry analysts do not publish research, or publish inaccurate or unfavorable research, about Holdco’s business, the price of Holdco Ordinary Shares and Holdco trading volume could decline. |
10
PART I
| ITEM 1. |
IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS |
Not applicable.
| ITEM 2. | OFFER STATISTICS AND EXPECTED TIMETABLE |
Not applicable.
| ITEM 3. | KEY INFORMATION |
| A. | [Reserved] |
| B. |
Capitalization and Indebtedness |
Not applicable.
| C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
| D. | Risk Factors |
Investing in our securities involves a high degree of risk. The risk factors described below disclose both material and other risks, and are not intended to be exhaustive and are not the only risks facing us. Additional risks not currently known to us or that we currently deem to be immaterial, or which are not identified because they are generally common to businesses, also may materially adversely affect our business, financial condition, results of operations and cash flows in future periods.
The occurrence of one or more of the events or circumstances described in these risk factors, alone or in combination with other events or circumstances, may have a material adverse effect on the business, financial condition, results of operations, cash flows and future prospects of Nuvo, in which event the market price of Holdco Ordinary Shares could decline, and you could lose part or all of your investment.
11
Risks Related to Our Business and Our INVU Platform
We are a development-stage company with a limited operating history. We may never be able to effectuate our business plan, achieve meaningful revenue or attain profitability.
We are a development-stage company and are subject to all of the risks inherent in the establishment of a new business enterprise. We have a limited operating history and only a preliminary and unproven business plan upon which investors may evaluate our prospects. We have not yet scaled commercial adoption of our INVU platform. Additionally, our INVU platform is currently cleared by the FDA for only limited monitoring capabilities, and the future commercial interest in our INVU platform, if any, will require FDA and other regulatory clearances or approvals for additional capabilities, and we may never obtain such clearances or approvals. Our ability to generate significant revenue from our operations and, ultimately, achieve profitability will depend on, among others things, whether we can commercialize our INVU platform as currently planned, whether we can complete the development of other features of our INVU platform, whether we can utilize the data we capture to make predictive recommendations and monetize these capabilities, our obtaining additional regulatory clearances, commercial adoption of our INVU platform, whether we can manufacture INVU on a commercial scale in such amounts and at such costs as we anticipate would be required to begin to achieve commercial success, and whether we can achieve market acceptance of our INVU platform and business model. We may never generate meaningful revenue or operate on a profitable basis. Even if we achieve profitability, we may not be able to sustain it.
It is difficult to predict our future revenues and appropriately budget for our expenses, and we have limited insight into trends that may emerge and affect our business. If actual results differ from our estimates or we adjust our estimates in future periods, our operating results and financial position could be materially affected.
We have a history of net losses, and we expect to continue to incur losses for the foreseeable future. If we ever achieve profitability, we may not be able to sustain it.
We have incurred losses since our inception, and we expect to continue to incur losses for the foreseeable future. For the years ended December 31, 2023, 2022 and 2021, we reported net losses of $33.655 million, $20.679 million and $34.512 million, respectively. As a result of these losses, as of December 31, 2023, 2022 and 2021, we had an accumulated deficit of $143.774 million, $110.119 million and $89.440 million, respectively. We expect to continue to incur significant sales and marketing expenses as we expand our sales and marketing efforts to increase adoption of our INVU platform, including through scaling our business in the United States and globally, expanding relationships with care providers, payer networks and strategic partners, and increasing awareness of our solutions among expectant mothers and their clinicians. In addition, we expect to continue to incur significant research and development and other expenses as we develop and utilize the measurements within our capabilities, expand our offerings by seeking clearance to provide some of these measurements to expectant mothers and clinicians, conduct additional clinical trials and studies on our INVU platform, and maintain and grow our intellectual property portfolio. In addition, we expect our general and administrative expenses to increase following the Business Combination due to the additional costs associated with being a public company. The net losses that we incur may fluctuate significantly from period to period. We will need to generate significant revenue and maintain or improve our gross margins to achieve and sustain profitability. Even if we achieve profitability, we may not remain profitable for any substantial period of time.
Our business model contemplates, among other things, an expansion of the approved uses for our INVU platform, proof to payers of reduced cost of delivering quality healthcare to expectant mothers, and additional collaborations with partners willing to recommend and prescribe the use of our INVU platform, all of which are subject to numerous risks and uncertainties and could result in the failure of our business model.
We have not yet demonstrated the feasibility of our INVU platform for commercial applications, including its ability to provide clinical-quality remote pregnancy care on a commercial scale. Currently, our INVU platform is cleared by the FDA to measure FHR, MHR and MUA during the antepartum period and the provision of remote NSTs. In addition, the ability to deliver MHR and FHR data is not necessarily novel and therefore may not enable us to gain or sustain a competitive advantage. Our business plan contemplates that our INVU platform ultimately provides monitoring for additional data and metrics. We may not be able to develop and utilize such additional measurements and include such measurements in our offerings, and even if we are able to do so, such data may not be of medical quality or equivalent to the data obtained from current standard of care. The expansion of our INVU platform’s usable capabilities, or the modification of our existing FDA cleared platform in response to feedback from third parties, such as medical professionals, also requires additional FDA clearance, which we may never receive, and any delay in receiving such clearance could also have a material adverse effect on our business. Additionally, we intend to expand globally and our INVU platform may be subject to the regulatory regimes of other non-U.S. jurisdictions, such as in Europe where we filed for a CE mark in March 2023 to offer NSTs using our FHR, MHR and MUA capabilities. Approval or clearance from the FDA, or comparable regulatory agency in other jurisdictions, to capture certain measurements and perform certain tests using our INVU platform, is not guaranteed and may take longer than planned. Also, regulatory approval in one jurisdiction does not mean that we will succeed in obtaining regulatory approval in other jurisdictions.
12
The software component of our INVU platform uses a cloud computing environment that processes and analyzes data and, ultimately, transmits personalized reports on maternal and fetal health metrics to the expectant mother and her clinician through digital visualization tools. The development of this cloud computing environment requires a considerable investment of technical, financial, and legal resources, which may not be available to us. It may also require separate regulatory clearances or approvals. Furthermore, it may not be technically viable for care providers and our partners to integrate the cloud with their businesses or platforms. There may also be public concerns regarding privacy and compliance with restrictive laws or regulations, including those with respect to management of health data, as well as concerns regarding hardware and software security and reliability issues associated with third-party mobile devices such as smartphones that would be used to access our cloud services.
Further, our business model contemplates the collection of a significant amount of personalized health data to develop a database sufficient for us to develop algorithms that may allow for effective and accurate predictive tools. We have yet to develop such a database, and we are not yet cleared to provide any such analytics, nor have we yet applied for or sought such clearances. Furthermore, even if we are able to develop such a database, we may not successfully develop effectively predictive algorithms. As a result, we may never ultimately develop our planned capabilities, or, if we do, care providers, expectant mothers or payers may not find such capabilities useful or cost effective.
The success of our business model also depends on our ability to:
| ● | generate widespread awareness, acceptance and adoption of our INVU platform and future products or services; |
| ● | prove out cost savings such that providers and payers clearly see value in the prescribing and use of our INVU platform; |
| ● | develop enhanced or new technologies or features that improve the convenience, efficiency, safety or perceived safety, and productivity of our INVU platform and future products or services, including the receipt of all regulatory clearances and approvals necessary for such enhanced or new technologies and features; |
| ● | significantly expand our commercial and strategic partnerships with enterprise-level entities in order to develop necessary product awareness and scale; |
| ● | properly identify customer needs and deliver new products or services or product enhancements to address those needs; |
| ● | obtain the regulatory approvals in a timely and cost-effective manner; attract and retain qualified personnel and collaborators; |
| ● | maintain quality control as we continue to commercialize our INVU platform; |
| ● | protect our inventions with patents or otherwise develop proprietary products and processes; and |
| ● | secure sufficient capital resources to expand both our continued research and development, and sales and marketing efforts. |
Given the foregoing, our success depends significantly upon, among other things, our ability to obtain additional regulatory approvals for our INVU platform’s more advanced capabilities and further expand such capabilities, materially expand our strategic partnerships to drive brand awareness and product usage, and prove that INVU reduces the cost of delivering quality healthcare for expectant mothers in order to help convince payers that INVU should be regularly prescribed and used. Our failure to successfully accomplish the foregoing could have a material adverse effect on our business, prospects, results of operation and financial position.
13
Our operating results may fluctuate from quarter to quarter, which makes our future results difficult to predict.
Our operating results and financial condition may fluctuate from quarter-to-quarter and year-to-year and are likely to vary due to a number of factors, many of which will not be within our control. Our operating results in any given quarter can be influenced by numerous factors, many of which are unpredictable or outside of our control, including:
| ● | the degree of market acceptance of our INVU platform and future products; |
| ● | our ability to compete with competitors and new entrants into our markets; |
| ● | the timing of our sales and deliveries of our INVU platform and future products to customers; |
| ● | changes in our pricing policies or those of our competitors, including our response to price competition; |
| ● | the effectiveness of our securing new orders and fulfilling existing orders; |
| ● | changes in the amount that we spend to develop and manufacture new products or technologies; |
| ● | changes in the amounts that we spend to promote our solutions; |
| ● | our ability to introduce new features and services and enhance our existing platform and our ability to generate significant revenue from new features and services and personalization to the products; |
| ● | our ability to respond to competitive developments, including pricing changes and the introduction of new products and services by our competitors; |
| ● | changes in the cost of satisfying our warranty obligations and servicing our products; |
| ● | litigation related expenses and/or liabilities; |
| ● | developments or disputes concerning our intellectual property or proprietary rights or our solutions, or third-party intellectual property or proprietary rights; |
| ● | fluctuations in currency exchange rates; |
| ● | general economic and political conditions and government regulations in the countries where we currently have significant numbers of systems, or where we currently operate or may expand in the future; and |
| ● | natural disasters, such as earthquakes, hurricanes, wildfires, and threats to public health, such as a resurgence of the COVID-19 pandemic. |
The impact of one or more of the foregoing and other factors may cause our operating results to vary significantly. As such, quarter-to-quarter comparisons of our operating results may not be meaningful and should not be relied upon as an indication of future performance. If we fail to meet or exceed the expectations of investors or securities analysts, the trading price of the Holdco Ordinary Shares could fall substantially, and we could face costly lawsuits, including securities class action suits.
14
Our business model contemplates a revenue model that is yet to be proven viable and is subject to numerous risks and uncertainties.
Our ability to generate significant revenue, and ultimately achieve profitability, will depend on securing commercial contracts on favorable economic terms. Currently, we have signed over a dozen commercial contracts with health systems, large private practice groups, and independent women’s health practices in the United States and Israel. We plan to focus on long-term enterprise level agreements with larger obstetrician-physician practice management groups and U.S. healthcare systems. We have entered into a strategic partnership with Philips primarily focused on providing a jointly integrated remote fetal monitoring solution targeted toward hospital networks in the U.S. However, we may not prove the benefits of our INVU platform, or such entities may not find our pricing to be attractive, either of which could cause our pricing model to fail. Ultimately, we aim to seek long-term contracts with payers, where we expect to receive revenue based, at least in part, on a percentage of cost-savings achieved by the applicable payers. We may not be able to develop a substantial body of data to prove to care providers and payers that the use of our INVU platform reduces medical care costs, and even if we are able to collect such data, we may not demonstrate cost savings, including as a result of the improvement of cost baseline in the long term, whether due to the success of our INVU platform or as other cost-effective offerings become available, or demonstrate improved quality of care and healthcare outcomes, in order to incentivize payers to encourage their obstetrician networks and expectant mothers to utilize our INVU platform. Our revenue model is also subject to many other factors, including the following:
| ● | payment models for remote healthcare solutions are still evolving, and the pricing arrangement we favor may not be accepted by care provider or payers; |
| ● | we may not be able to find a sufficient number of implementers to stimulate market interest or reach the scale necessary to make our INVU platform a cost-effective solution, which is a key factor for acceptance by care providers and ultimately the payers; |
| ● | even if we can demonstrate cost savings from use of our INVU platform, we may be unable to secure arrangements with payers that share any cost-saving with us, on favorable terms to us or at all; |
| ● | we may not be able to secure meaningful up-front and ongoing payments; |
| ● | contracted payment terms will likely vary among counterparties, making it difficult to predict revenues; |
| ● | manufacturing or maintenance costs may be higher than expected and we may not be able to adjust our pricing model to accommodate for these increases, which will increase our operating expenses and reduce our margins; and |
| ● | we may not be able to accumulate sufficient data of the type and quality we need to develop predictive tools, and even if we are able to do so, we may not be successful in generating revenue from these tools. |
Manufacturers of medical devices have a history of price competition, and we may not be able to achieve or maintain satisfactory pricing for our INVU platform. If we are forced to lower the price we charge for our INVU platform, our gross margins will decrease, which will harm our ability to invest in and grow our business. If we are unable to maintain our prices, or if our costs increase and we are unable to offset such increase with an increase in our prices, our margins could erode. We may be subject to significant pricing pressure, which could harm our business and results of operations. Any of these risks and uncertainties could cause our revenue model to fail.
Our success depends in large part on our ability to develop, market and sell our INVU platform. If we are unable to successfully develop, market and sell this product, our business prospects will be significantly harmed and we may be unable to achieve revenue growth or profitability.
Our future financial success will depend substantially on our ability to further development, and effectively and profitably market and sell, our INVU platform. Our products may not gain market acceptance in the United States or internationally or otherwise attain and maintain any level of market share.
15
The commercial success of our INVU platform and any of our planned or future products will depend on a number of factors, including, but not limited to, the following:
| ● | the actual and perceived effectiveness, safety and reliability, and clinical benefit, of our INVU platform, especially relative to the current standard of care obtained within healthcare facilities; |
| ● | the degree to which expectant mothers, care providers, such as large healthcare systems obstetrician- physician practice management groups, and payer networks adopt and continue to use and prescribe our INVU platform; |
| ● | the degree to which expectant mothers use our INVU platform correctly and consider it a valuable tool during their pregnancies; |
| ● | the availability, relative cost and perceived advantages and disadvantages of alternative technologies for pregnancy monitoring; |
| ● | the results of additional clinical and other studies relating to the health, safety, economic or other benefits of our INVU platform; |
| ● | whether key thought leaders in the medical community adopt our INVU platform over alternatives and products offered by our competitors, and the extent to which we are successful in educating physicians and healthcare providers about the benefits of our INVU platform; |
| ● | the success of our strategic partnerships and our current and future strategic partners; |
| ● | our ability to successfully market, sell and distribute our INVU platform and any related platform products, including, without limitation, any of our planned cloud-based solutions derived from the data we expect to collect from expectant mothers, including our plan to identify patterns and trends associated with certain risks and outcomes from which we may derive predictive recommendations that could be useful to individual expectant mothers; |
| ● | our reputation among care providers, such as obstetrician-physician management groups; |
| ● | our ability to obtain, maintain, protect and enforce our intellectual property rights in and to our INVU platform; |
| ● | our ability to maintain compliance with all regulatory requirements applicable to our INVU platform; and |
| ● | our ability to continue to maintain quality control and real-time data processing ability as we continue to commercialize our INVU platform. |
If we fail to successfully market and sell our products cost-effectively and develop, maintain and expand our market share, we will not be able to achieve profitability, which will harm our business, financial condition and results of operations. Our ability to grow our revenue in future periods will depend on our ability to successfully penetrate our target markets and increase sales of our product, which will, in turn, depend in part on our success in driving adoption and increased use of our products as well as the prices we can charge.
We are highly dependent on the successful development, marketing and sale of our INVU platform and the related products and services.
Our INVU platform comprises the basis of our business. As a result, the success of our business plan is highly dependent on our ability to develop, manufacture and commercialize our INVU platform and related products and services, and our failure to do so could cause our business to fail. Successful commercialization of medical devices, such as our INVU platform, is a complex and uncertain process, dependent on the efforts of management, manufacturers, medical professionals, third-party payers, our strategic partners, as well as general economic conditions, among other factors. Any factor that adversely impacts the development and commercialization of our INVU platform will have a negative impact on our business, financial condition, results of operations and prospects. Some potential factors include:
16
| ● | our ability to significantly scale our pregnancy care population, together with the necessary increase in manufacturing capacity that would be required to produce the hardware components of our INVU platform to serve a much larger population of expectant mothers; |
| ● | our ability to adapt our INVU platform to the extent necessary to work for a substantial majority of expectant mothers; |
| ● | our ability to achieve sufficient market acceptance by expectant mothers, strategic partners, commercial customers, and other medical and clinical professionals, third-party payers and others in the medical community; |
| ● | our ability to compete with existing pregnancy care solutions, such as currently standard in-person, non-remote, monitoring solutions and current or future competing remote solutions; |
| ● | our ability to establish, maintain and expand our sales, marketing and distribution networks, such as the Philips distribution channel; |
| ● | our ability to obtain or maintain necessary regulatory approvals, including with respect to any changes to our products based upon feedback from third parties such as medical professionals; and |
| ● | our ability to effectively protect our intellectual property. |
Our inability to successfully obtain clearance or approval for and subsequently commercialize our INVU platform or related products and services would have a material adverse effect on our business, financial condition, results of operations and prospects.
Our commercial success will depend upon attaining significant market acceptance of our INVU platform among expectant mothers, care providers, payers and others in the medical community. If we are unable to successfully achieve substantial market acceptance and adoption of our INVU platform, our business, financial condition and results of operations would be harmed.
Our commercial success will depend in large part on the acceptance of our INVU platform by expectant mothers, care providers, payers and others in the medical community as safe for both an expectant mother and her unborn baby, useful and cost-effective. We cannot predict how quickly, if at all, care providers, such as obstetrician-physician practice management groups, hospitals and healthcare systems, and payers will accept our INVU platform. These participants may not readily accept our INVU platform over current standard of care obtained within healthcare facilities or competing products or alternatives in the near term or at all. Additionally, expectant mothers may prefer the current standard of care, including in-office visits during which they have the in-person attention of a medical professional. Further, some expectant mothers may be unwilling to use our INVU platform given that it represents new technology without a significant history of use and results. Care providers, or value analysis committees at their hospitals, as well as third-party payers, may also perceive our products to be too costly, or may believe that the benefits of our INVU platform and results from clinical trials, such as relative ease of use, are not sufficiently greater than other alternatives to justify our INVU platform’s pricing. This perception may continue to be heightened due to any budgetary and financial constraints faced by care providers, including hospitals and other facilities. Moreover, the medical community may be unwilling to depart from the current standard of care for pregnancy monitoring and pregnancy care management. Medical professionals tend to be slow to change their medical diagnostic practices because of perceived liability risks arising from the use of new technology or products, and they may not recommend our INVU platform or other products integrated with our technology until there is long-term clinical evidence to convince them to alter or modify their existing pregnancy monitoring methods. The use of wearable technology, artificial intelligence, machine learning and other technology-based platforms to provide pregnancy monitoring and care management is a recent phenomenon, and therefore, our INVU platform may not become broadly accepted by physicians, patients, hospitals and others in the medical community, even if it is approved by the appropriate regulatory authorities for marketing and sale. Our efforts to educate expectant mothers, care providers, payers and others in the medical community on the benefits of our INVU platform require significant resources and may not be successful. Our efforts to educate the marketplace may require more resources than are required by conventional technologies marketed by our competitors. Moreover, in the event that our INVU platform or other products integrated with our technology are the subject of guidelines, clinical studies or scientific publications that are unfavorable or damaging, or otherwise call into question its benefits. Our ability to grow sales of our INVU platform and drive market acceptance will depend on successfully educating expectant mothers, care providers, such as obstetrician-physician practice management groups, payers and others in the medical community of the relative benefits of our INVU platform and its cost-effectiveness.
17
The degree of market acceptance by both care providers and expectant mothers of our INVU platform will depend on a number of additional factors, including:
| ● | regulatory requirements regarding product labeling or product inserts; |
| ● | limitations or warnings contained in the labeling cleared or approved by the FDA or other regulatory authorities; |
| ● | the existence of current in-person monitoring for expectant mothers, including that certain expectant mothers may prefer in-person care by a medical professional; |
| ● | coverage determinations and reimbursement levels of third party payers; |
| ● | pricing and cost of our INVU platform in relation to alternative products and methods; |
| ● | timing of market introduction of competing products and the sales and marketing initiatives of such products; |
| ● | the access to, ease of use, stability of device performance and error rate of our INVU platform by both care providers and expectant mothers relative to alternative products and methods; |
| ● | the willingness and ability of expectant mothers to adopt new technology, including its perceived safety and ease of use; |
| ● | our ability to provide incremental clinical and economic data that show the safety, clinical efficacy and cost-effectiveness of, and benefits from, our INVU platform; and |
| ● | the effectiveness of our sales and marketing efforts for our INVU platform. |
If we are unable to successfully achieve substantial market acceptance and adoption of our INVU platform, our business, financial condition and results of operations would be harmed. Even if our INVU platform achieves market acceptance, it may not maintain that market acceptance over time if competing products or technologies, which are more cost effective or received more favorably, are introduced. Failure to achieve or maintain market acceptance or market share would limit our ability to generate revenue and would significantly harm our business, financial condition and results of operations.
We currently have a limited sales and marketing organization. If we are unable to develop our sales and marketing capability on our own or through collaborations with marketing partners, we will not be successful in commercializing our INVU platform.
Currently, our sales and marketing team consists of our VP Marketing, Product Specialist and our business development team in Israel and, as a result, we have no meaningful marketing and sales capabilities. We intend to sell our INVU platform primarily to and through our implementers in the near term, and ultimately through third-party payers. We also intend to utilize the data we capture to make predictive recommendations and monetize these capabilities. However, we may not be successful in doing so. To the extent that we enter into co-promotion or other licensing arrangements, our INVU platform revenue is likely to be lower than if we directly marketed or sold our INVU platform. In addition, any revenue we receive will depend in whole or in part upon the efforts of such third parties, which may not be successful and are generally not within our control. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our INVU platform. If we are not successful in commercializing our INVU platform, either on our own or through collaborations with one or more third parties, our future revenue will suffer and we may incur significant additional losses.
18
The success of our business may be dependent on our strategic partnerships and collaborations.
Strategic relationships with our implementers and validators are and will be important to the success of our business. See Item 4.B. “Business Overview—Sales and Marketing.” We anticipate deriving a significant portion of revenues in the near term from our implementers, which are provider partners with an installed base of clinicians that understand how to prescribe and use our INVU platform for the expectant mothers they care for. We currently have over a dozen enterprise-level agreements, and our future success depends on our ability to enter into such agreements with additional implementers. Our prospects also depend on our validators to build robust clinical evidence based on our already developed INVU platform, as well as research experts, mainly academic centers, to analyze our data signals and help determine predictive markers through such data. Our strategic partners may have the right to abandon the use of our INVU platform and terminate applicable agreements, including payment obligations, prior to or upon the expiration of the agreed-upon agreement terms. We may not be successful in establishing strategic partnerships or collaborative arrangements on acceptable terms or at all, our collaborative partners may terminate any such agreements prior to their stated terms, our collaborative arrangements may not result in successful product development, validation or commercialization and we may not derive any revenues from such arrangements. If we do not successfully develop and maintain strategic partnerships or collaborative arrangements, our business, financial condition and results of operations would be materially and adversely affected.
Any strategic partnerships or collaborative arrangements that we have established or may establish in the future may not be successful or we may otherwise not realize the anticipated benefits from these strategic partnerships or collaborations. We do not control third parties with whom we have or may have strategic partnerships or collaborative arrangements, and we will rely on them to achieve results which may be significant to us. In addition, any current or future strategic partnerships or collaborative arrangements may place the development and commercialization of our technology outside our control, may require us to relinquish important rights or may otherwise be on terms unfavorable to us.
We have entered into certain, and expect to enter into additional, strategic partnerships or collaborative arrangements with respect to the development, validation and commercialization of our INVU platform with different relevant industry participants, including our implementers and validators. Any future potential strategic partnerships or collaborative arrangements may require us to rely on external consultants, advisors and experts for assistance in several key functions, including research and development, manufacturing, regulatory, intellectual property, commercialization and distribution. We cannot and will not control these third parties, but we may rely on them to achieve results, which may be significant to us. Relying upon these strategic partnerships or collaborative arrangements subjects us to a number of risks, including:
| ● | we may not be able to control the amount and timing of resources that our partners or collaborators may devote to our technology; |
| ● | should a partner or collaborator fail to comply with applicable laws, rules or regulations when performing services for us, we could be held liable for such violations; |
| ● | we may be required to relinquish important rights, such as marketing and distribution rights, including the ability to distribute to hospital networks without Philips in accordance with the exclusivity terms set by the Philips MPA, pursuant to meeting certain sales targets; |
| ● | business combinations or significant changes in a partner or collaborator’s business strategy may adversely affect such person’s willingness or ability to complete its obligations under any arrangement; |
| ● | our partners or collaborators may default on their payments to us or fail to deliver standby letters of credit or financial guarantees, and it may be time consuming and difficult to enforce such payment obligations and obligations to provide standby letters of credit and financial guarantees in various jurisdictions, and we may be unsuccessful in enforcing such obligations; |
19
| ● | our current or future partners or collaborators may utilize our proprietary information in a way that could expose us to competitive harm; |
| ● | our partners or collaborators could obtain ownership or other control over intellectual property that is material to our business, or we may be required to jointly own certain of our intellectual property with such third parties; and |
| ● | strategic partnerships or collaborative arrangements are often terminated or allowed to expire or remain unformalized by a written agreement, which could delay the ability to commercialize our technology. |
In addition, if disputes arise between us and any of our partners or collaborators, it could result in the delay or termination of the development, validation or commercialization of products containing our technology, lead to protracted and costly legal proceedings, or cause partners or collaborators to act in their own interest, which may not be in our interest. As a result, the strategic partnerships or collaborative arrangements that we have entered into or may enter into may not achieve their intended goals.
If any of these scenarios materialize, they could have a material adverse effect on our business, financial condition, results of operations and prospects.
The audited consolidated financial statements for the year ended December 31, 2023, include an explanatory paragraph in our independent registered public accounting firm’s audit report stating that there are conditions that raise substantial doubt about our ability to continue as a going concern, and we will need to obtain additional financing to fund our future operations and continue as a going concern. If we are unable to obtain such financing, we may be unable to complete the development and commercialization of our INVU platform.
Our operations have consumed substantial amounts of cash since inception. Our net losses were $33.655 million, $20.679 million and $34.512 million for the years ended December 31, 2023, 2022 and 2021, respectively. As of December 31, 2023, our primary sources of liquidity were cash and cash equivalents totaling $0.6 million. We anticipate that our future cash requirements will continue to be significant. As a result of the above, in connection with our assessment of going concern considerations performed in connection with the audit for the year ended December 31, 2023, management has determined that our liquidity condition raises substantial doubt about our ability to continue as a going concern through twelve months from the date those audited consolidated financial statements are available to be issued. Additionally, the opinion of our independent registered accountants on our audited financial statements included in this Annual Report contains an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern. Our ability to continue operations as a going concern will depend on, among other things, our ability to obtain funding through equity and/or debt financing, potential partnership arrangements, sale of products, as well as our ability to manage our expenses. While we believe our strategies will generate funding that will be sufficient to continue as a going concern, if these strategies are unsuccessful, then we may need to realize assets and extinguish liabilities other than in the ordinary course of business and at amounts different to those disclosed in our financial statements. Our financial statements do not contain any adjustments to the amounts or classifications of recorded assets or liabilities that might be necessary if we do not continue as a going concern. The financial statements take no account of the consequences, if any, of the effects of unsuccessful product development or commercialization, nor of any inability of our company to obtain adequate funding in the future. We expect that we will need to obtain additional financing to implement our business plan as described in this Annual Report. Such financing could include equity financing, which may be dilutive to shareholders, or debt financing, which would likely restrict our ability to borrow from other sources. In addition, such securities may contain rights, preferences or privileges senior to those of the rights of our current shareholders. Additional funds may not be available when we need them, on terms attractive to us, or at all. If adequate funds are not available on a timely basis, we may be required to curtail the development of our INVU platform and related products or services, or materially delay, curtail, reduce or terminate our research and development and commercialization activities. We could be forced to sell or dispose of our rights or assets. Any inability to raise adequate funds on commercially reasonable terms could have a material adverse effect on our business, financial condition, results of operation and prospects, including the possibility that a lack of funds could cause our business to fail and liquidate with little or no return to investors.
20
The manufacturing and supply of our INVU platform is subject to various factors outside our direct control, including those related to our dependence on third-party manufacturers and suppliers, which could harm our business, financial condition and results of operations.
In 2019, we began a hybrid production process, involving both in-house assembly of our wearable wireless sensor band and the use of sub-contractors for the supply and production of the component elements. In 2021, we started to fully outsource our manufacturing operation for our first production batch to Flextronics Medical Sales and Marketing, Ltd., a company located in Israel, and we currently anticipate continuing to do so for all future production batches. Pursuant to our manufacturing plan, our printed circuit boards, or PCBs, are manufactured in China and Israel and fabricated in China, acoustic sensors are sourced from Japan, reusable ECG sensors are sourced from China and accessories are sourced from Israel and the United States. The products are then shipped to Israel where they are assembled into a complete sensor band. Accessories are added in the United States and the product is packaged and ready for delivery to the expectant mother. While the foregoing manufacturing and supplier relationships are adequate for our current operations, our successful growth will require that we either expand our existing manufacturing and supplier relationships or enter into new relationships, which we may not be able to do on a commercially reasonable basis or at all. We do not have significant experience with scalable manufacturing, and we expect to remain dependent for the foreseeable future on third parties. Given our dependence on third-party manufacturers and suppliers, we are subject to additional risks relating to these third parties, including: insufficient capacity or delays in meeting our demand (including due to any problems with our third-party manufacturers’ and suppliers’ respective supply chains); inadequate manufacturing yields, inferior quality and excessive costs; inability to manufacture products that meet the agreed upon specifications; inability to obtain an adequate supply of materials; inability to comply with the relevant regulatory requirements for the manufacturing process; limited warranties on products supplied to us; inability or failure to comply with our contractual obligations; potential increases in prices; and increased exposure to potential misappropriation of our intellectual property. Additionally, we currently do not have immediate contingency arrangements if one of our primary suppliers or manufacturers became unable to meet our product demand, including, without limitation, due to international shipping delays, whether due to trade embargo issues, weather-related delays or otherwise.
The manufacture and supply of our INVU platform, both in-house and by our third-party manufacturing and supply partners, in compliance with ISO standards and the FDA’s regulations requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. We and our third-party manufacturers and suppliers may encounter difficulties in production, including difficulties with production costs and yields, quality control, quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced FDA requirements, other federal and state regulatory requirements, as well as foreign regulations. If we fail to manufacture our INVU platform in compliance with ISO standards and the FDA’s regulations, or if the manufacturing facilities suffer disruptions, machine failures, slowdowns or disrepair, we may not be able to fulfill customer demand and our business would be harmed. Further, we do not expect to maintain excess product inventory on hand and intend to manufacture our INVU platform using near term demand forecasts and customer orders. As a result, deviations from our forecasts or large unexpected customer orders may result in delays in fulfilling customer orders, which would cause customer dissatisfaction and may harm our reputation. Finally, failure to comply with local laws, regulations and standards, in the countries in which our manufacturing facilities are located, which may be outside of our control, may subject us to legal and regulatory scrutiny, proceedings and penalties from such outside authorities.
A majority of our software development team is based in Ukraine and our business could be harmed if political instability or military conflict disrupts our team’s ability to operate.
In February 2022, Russia invaded Ukraine and global sanctions were announced by the U.S., European Union, Japan and additional countries against Russia, which caused disruptions in the energy, metal and other commodities supply chain and increased costs. Although the length and impact of the ongoing military conflict is highly unpredictable, the conflict in Ukraine could lead to market disruptions, including significant volatility in commodity prices, credit and capital markets, as well as supply chain interruptions.
We are actively monitoring the situation in Ukraine and will continue to assess any impact it may have on our business. We have a team of software engineers based in Ukraine that are responsible for a portion of our engineering and software development initiatives. If this team’s operations were disrupted or discontinued due to local instability or political, economic or military conditions, then our ability to provide services to some of our current customers and the development of new products or enhancement of existing products could be delayed, and our results of operations could be adversely affected. While the conflict in Ukraine has not yet had any material impact on our business to date, there is no way of predicting the progress or outcome of the conflict as it continues to rapidly develop and is out of our control.
21
Holdco will incur increased costs as a result of operating as a public company.
As a public company, Holdco will incur significant legal, accounting and other expenses that Nuvo did not incur as a private company. As a public company, Holdco is subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, as well as rules adopted, and to be adopted, by the SEC and Nasdaq. Holdco’s management and other personnel need to devote a substantial amount of time to these compliance initiatives and may not effectively or efficiently manage the transition into a public company. Moreover, Holdco expects these rules and regulations to substantially increase its legal and financial compliance costs and to make some activities more time-consuming and costly. For example, such rules and regulations may make it more difficult and expensive for Holdco to obtain director and officer liability insurance and Holdco may be forced to accept reduced policy limits or incur substantially higher costs to maintain the same or similar coverage. Holdco cannot predict or estimate the amount or timing of additional costs it may incur to respond to these requirements. The impact of these requirements could also make it more difficult for Holdco to attract and retain qualified persons to serve on the Holdco Board, its Board committees or as executive officers.
Most members of our management team have limited experience managing a publicly traded company, interacting with public company investors and complying with the increasingly complex laws pertaining to public companies in the United States. The additional demands associated with being a public company may disrupt regular operations of Holdco’s business by diverting the attention of some of its senior management team away from revenue producing activities to management and administrative oversight, adversely affecting Holdco’s ability to attract and complete business opportunities and increasing the difficulty in both retaining professionals and managing and growing its businesses. Holdco’s management team may not successfully or efficiently manage its transition to being a public company subject to significant regulatory oversight and reporting obligations under the U.S. federal securities law and the continuous scrutiny of securities analysts and investors.
In addition, the public reporting obligations associated with being a public company in the United States may subject Holdco to litigation as a result of increased scrutiny of its financial reporting. If Holdco is involved in litigation regarding its public reporting obligations, this could subject Holdco to substantial costs, divert resources and management attention from Holdco’s business and seriously undermine Holdco’s business.
Any of these effects could harm Holdco’s business, financial condition and results of operations.
Our business is subject to the risks associated with doing business in China.
As a result of our reliance on third-party suppliers located in China, our results of operations, financial condition, and prospects are subject to a certain degree to economic, political and legal developments in China, including government control over capital investments or changes in tax regulations that are applicable to us. China’s economy differs from the economies of most developed countries in many respects, including with respect to the amount of government involvement, level of development, growth rate and control of foreign exchange, and allocation of resources. Since we rely on certain suppliers located in China for certain parts, our business is subject to the risks associated with doing business in China, including:
| ● | trade protections measures, such as tariff increases, and import and export licensing and control requirements; |
| ● | potentially negative consequences from changes in tax laws; |
| ● | difficulties associated with the Chinese legal system, including increased costs and uncertainties associated with enforcing contractual obligations in China; |
| ● | historically lower protection of intellectual property rights; |
| ● | changes and volatility in currency exchange rates; and |
| ● | unexpected or unfavorable changes in regulatory requirements. |
The United States and China historically had a complex relationship that has included actions that have impacted trade between the two countries and globally. If trade relations with the United States were to result in trade restrictions, if social or political unrest were to disrupt business in China, or if other events in China significantly reduced or disrupted business activities in China, that may materially and adversely harm our business.
22
If we fail to grow or optimize our sales and marketing capabilities and develop widespread brand awareness cost-effectively, our growth will be impeded and our business may suffer.
We intend to commercialize our INVU platform and grow brand awareness, commencing in the United States, by establishing a network of implementers and validators, and strategically working with distribution partners such as Philips. We may also expand our presence in international territories in the future, with the goal of becoming a global leader in pregnancy solutions from the first days of pregnancy onward. We plan to take a measured approach to expand and optimize our sales infrastructure to grow our customer base and our business. In developing a U.S. team, identifying and recruiting qualified personnel and training them on the use of our INVU platform, on applicable federal and state laws and regulations and on our internal policies and procedures, will require significant time, expense and attention. It may take significant time before our sales representatives are fully trained and productive. In particular, if we are unable to hire, develop and retain talented sales personnel or if new sales personnel are unable to achieve desired productivity levels in a reasonable period of time, we may not be able to realize the expected benefits of this investment or increase our revenue. Our business may be harmed if our efforts to expand either fail to generate a corresponding increase in revenue or otherwise result in a decrease in our operating margin.
We plan to dedicate significant financial and other resources to our marketing programs, particularly as we grow our sales territories, which may require us to incur significant upfront costs, such as in connection with care provider training seminars and sessions and relevant content generation and promotion.
In addition, we believe that developing and maintaining awareness of our INVU platform and the impact it has on providers, patients, and payers in a cost-effective manner is critical to achieving broad acceptance of our INVU platform and attracting new provider groups and expectant mothers. Brand promotion activities, such as advertising, social media and other communication channels, may not generate awareness or increase revenue and, even if they do, any increase in revenue may not offset the costs and expenses we incur in building our brand. If we fail to successfully promote, maintain and protect our brand, we may fail to attract or retain the care providers and expectant mothers necessary to realize a sufficient return on our brand-building efforts, or to achieve the widespread brand awareness that is critical for broad customer adoption of our INVU platform.
We plan to do business globally, including in certain countries in which we may have limited resources and would be subject to additional regulatory burdens and other risks and uncertainties.
We expect to do business globally, currently including North America and certain countries in Europe. Commercialization of our INVU platform in foreign markets, either directly or through third parties, is subject to additional risks and uncertainties, including:
| ● | reimbursement and insurance coverage; |
| ● | our inability to find strategic partners, dealers or distributors in specific countries or regions; |
| ● | our inability to directly control commercial activities of third parties; |
| ● | our limited resources to be deployed to a specific jurisdiction; |
| ● | the burden of complying with complex and changing regulatory, tax, accounting and legal requirements; |
| ● | different clinical practice and customs in foreign countries affecting acceptance of our INVU platform in the marketplace; |
| ● | import or export licensing and other requirements; |
23
| ● | longer accounts receivable collection times; |
| ● | longer lead times for shipping; |
| ● | language barriers for technical training; |
| ● | reduced protection of intellectual property rights in some foreign countries; |
| ● | foreign currency exchange rate fluctuations; and |
| ● | interpretations of contractual provisions governed by foreign laws in the event of a contract dispute. |
Specifically, we are or may be subject to the U.S. Foreign Corrupt Practices Act of 1977, as amended, or FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, the United Kingdom Bribery Act 2010, the Proceeds of Crime Act 2002, Chapter 9 (sub-chapter 5) of the Israeli Penal Law, 1977, the Israeli Prohibition on Money Laundering Law–2000 and possibly other anti-bribery and anti-money laundering laws in countries outside of the United States in which we conduct our activities. As we engage in business in certain countries, we and our agents and independent contractors may have direct or indirect interactions with officials and employees of government agencies or state-owned or affiliated entities. We may be held liable for the corrupt or other illegal activities of these third-party business partners and intermediaries, our employees, representatives, contractors, partners and agents, even if we do not explicitly authorize such activities. As we expand our international business, our risks under these laws may increase, including that we may become subject to government actions against, fines, penalties and resultant reputational harm, any of which could have a material adverse effect on our business, financial position and results of operations.
We are highly dependent on our senior management team and key personnel, as well as other employees, and our business could be harmed if we are unable to attract and retain personnel necessary for our success.
We are highly dependent on our senior management team and directors and key personnel (many of whom are seasoned medical device professionals with a wide array of experience, such as women’s health, medical technology and healthcare), as well as other employees. Our success will depend on our ability to retain senior management and to attract and retain qualified personnel in the future, including sales and marketing professionals, engineers and other highly skilled personnel and to integrate current and additional personnel in all departments. The loss of members of our senior management, sales and marketing professionals and engineers as well as contract employees at our manufacturing facilities could result in delays in product development and harm our business. If we are not successful in attracting and retaining highly qualified personnel, it would have a negative impact on our business, financial condition and results of operations. At the end of February 2024, we experienced a substantial change in our management team, with the departure of our current Chief Executive Officer, Kelly Londy, who accepted a leading position at a non-competitive multinational healthcare company. Ms. Londy remains involved with Nuvo as a member of Nuvo’s Strategic Advisory Council. While Robert Powell, a member of our board of directors with extensive experience in the healthcare industry, assumed the role of Chief Executive Officer and while we believe we will continue to have a strong experienced leadership team, leadership changes can be inherently difficult to manage and may cause disruption to our business. Our business may be disrupted if we cannot successfully integrate our new Chief Executive Officer or we lose the services of any other members of senior management team and directors and key personnel.
Competition for skilled personnel in our market is intense and may limit our ability to hire and retain highly qualified personnel on acceptable terms, or at all. Despite our efforts to retain valuable employees, members of our management and development teams may terminate their employment with us on short notice.
24
Our industry is highly competitive and is subject to technological change, which may result in new products or solutions that are superior to our INVU platform or other future products we may bring to market from time to time. If we are unable to anticipate or keep pace with changes in the marketplace and the direction of technological innovation and customer demands, our technology may become less useful or obsolete and our operating results will suffer.
The pregnancy monitoring and management industry is rapidly evolving and subject to intense and increasing competition. To compete successfully and to be able to establish and maintain a competitive position in current and future technologies, we will need to demonstrate the advantages of our technology, specifically our INVU platform, over currently well-established alternative solutions, such as conventional in-person monitoring at an obstetrician’s office, in a hospital or at another healthcare facility. There are currently a number of existing monitoring devices that strive to provide rich and robust data, all of which are also limited to use within healthcare facilities. These include Monica Healthcare, now part of General Electric, Nemo Healthcare and Philips Avalon CL, and their respective competitive technologies and devices all have received regulatory approval for the intrapartum period for singleton pregnancies at healthcare facilities by medical professionals. There are also devices that seek to provide distributed care and generally work remotely, such as Sense4Baby, Pregnabit, Bloom, and Heramed. Furthermore, as the market expands, we expect the entry of additional competitors, such as cloud computing companies or leading IT companies, who may have longer operating histories, more extensive international operations, greater name recognition, and substantially greater technical, marketing and financial resources. If our technology is not, or our future products or services are not, competitive based on these or other factors, our business would be harmed.
Under applicable employment and anti-competition laws, we may not be able to enforce covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees.
We generally enter into non-competition agreements with our employees. These agreements prohibit our employees from competing directly with us or working for our competitors or customers for a limited period after they cease working for us. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees work and it may be difficult for us to restrict our competitors from benefiting from the expertise that our former employees or consultants developed while working for us. For example, in Israel, where most of our employees are based, Israeli labor courts have required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the protection of a company’s trade secrets or other proprietary knowhow, and thus non-competition agreements with employees are generally unenforceable after termination of employment.
Environmental, social and corporate governance (“ESG”) issues, including those related to climate change and sustainability, may have an adverse effect on our business, financial condition and results of operations and damage our reputation.
There is an increasing focus from certain investors, customers, consumers, employees and other stakeholders concerning ESG matters. Additionally, public interest and legislative pressure related to public companies’ ESG practices continue to grow. If our ESG practices fail to meet regulatory requirements or investor, customer, consumer, employee or other shareholders’ evolving expectations and standards for responsible corporate citizenship in areas including environmental stewardship, support for local communities, board of directors and employee diversity, human capital management, employee health and safety practices, product quality, supply chain management, corporate governance and transparency, our reputation, brand and employee retention may be negatively impacted, and our customers and suppliers may be unwilling to continue to do business with us.
Customers, consumers, investors and other shareholders are increasingly focusing on environmental issues, including climate change, energy and water use, plastic waste and other sustainability concerns. Concern over climate change may result in new or increased legal and regulatory requirements to reduce or mitigate impacts to the environment. Changing customer and consumer preferences or increased regulatory requirements may result in increased demands or requirements regarding plastics and packaging materials, including single-use and non-recyclable plastic products and packaging, other components of our products and their environmental impact on sustainability, or increased customer and consumer concerns or perceptions (whether accurate or inaccurate) regarding the effects of substances present in certain of our products. Complying with these demands or requirements could cause us to incur additional manufacturing, operating or product development costs.
If we do not adapt to or comply with new regulations, including the SEC’s published proposed rules that would require companies to provide significantly expanded climate-related disclosures in their periodic reporting, which may require us to incur significant additional costs to comply and impose increased oversight obligations on our management and board of directors, or fail to meet evolving investor, industry or stakeholder expectations and concerns regarding ESG issues, investors may reconsider their capital investment in our Company, we may become subject to penalties, and customers and consumers may choose to stop purchasing our products, if approved for commercialization, which could have a material adverse effect on our reputation, business or financial condition.
25
Failure to maintain the security and functionality of our information systems, or to defend against or otherwise prevent a cybersecurity attack or data breach, could adversely affect our business, financial position, results of operations and liquidity.
We depend on our information technology systems for the efficient functioning of our business, including the manufacture, distribution and maintenance of our INVU platform, as well as for purchasing and inventory management. We also collect, store, use, retain, disclose, transfer and otherwise process a significant amount of confidential, sensitive and personal information from and about the expectant mothers that we care for and our employees, including tax information, health information and payroll data. In addition to internal resources, we rely on third party service providers in providing our services, including to provide continual maintenance and enhancements and security of any protected data. Such third-party service providers have access to confidential, sensitive and personal information about the expectant mothers we care for and employees, and some of these service providers in turn subcontract with other third-party service providers. Through contractual provisions and third-party risk management processes, we take steps to require that our service providers, and their subcontractors, protect our confidential, sensitive and personal information. However, due to the size and complexity of our technology platform and services, the amount of confidential, sensitive and personal information that we store and the number of expectant mothers, employees and third-party service providers with access to confidential, sensitive and personal information, we are vulnerable to a variety of intentional and inadvertent cybersecurity attacks and other security-related incidents and threats, which could result in legal risks, enforcement actions, fines, reputational damages and a material adverse effect on our business, financial position, results of operations and liquidity. Technological interruptions would disrupt our operations, including our ability to timely ship and track product orders, project inventory requirements, manage our supply chain and otherwise adequately service providers or expectant mothers or disrupt their ability to use our INVU platform.
Threats to our information technology systems and data security can take a variety of forms. Hackers may develop and deploy viruses, worms and other malicious software programs that attack our networks and data centers or those of our service providers. Additionally, unauthorized parties may attempt to gain access to our systems or facilities, or those of third parties with whom we do business, through fraud, trickery, or other forms of deceiving our employees or contractors, direct social engineering, phishing, credential stuffing, ransomware, denial or degradation of service attacks and similar types of attacks against any or all of us, the expectant mothers we care for and our service providers. Other threats include inadvertent security breaches or theft, misuse, unauthorized access or other improper actions by our employees, expectant mothers we care for, service providers and other business partners. Cybersecurity attacks and other security-related incidents are increasing in frequency and evolving in nature. Since the beginning of the war between Israel and Hamas which began on October 7, 2023, Israeli and Israeli associated companies have become more frequently the target of cyberattacks. As such, the risk of a cyberattack against our information technology systems and data security may become heightened.
We have implemented policy, procedural, technical, physical and administrative controls with the aim of protecting our networks, applications, bank accounts, and the confidential, sensitive and personal information entrusted to us from such threats. However, given the unpredictability of the timing, nature and scope of cybersecurity attacks and other security related incidents, our technology may fail to adequately secure the confidential health information and personally identifiable information we maintain in our databases and security procedures and controls that we or our service providers have implemented may not be sufficient to prevent such incidents from occurring. Furthermore, because the methods of attack and deception change frequently, are increasingly complex and sophisticated, and can originate from a wide variety of sources, including third parties such as service providers and even nation-state actors, it is possible that we may not be able to anticipate, detect, appropriately react and respond to, or implement effective preventative measures against, all cybersecurity attacks and other security-related incidents. As a result, our business, financial condition, results of operations and liquidity could be materially and adversely affected.
The occurrence of any actual or attempted cybersecurity attack or other security-related incident or a data breach, the reporting of such incidents, whether accurate or not, or our failure to make adequate or timely disclosures to the public or law enforcement agencies following any such event, whether due to delayed discovery or a failure to follow existing protocols, could result in liability to the expectant mothers we care for and/or regulators, which could result in significant fines, litigation penalties, orders, sanctions, adverse publicity, litigation or actions against us or our service providers by governmental bodies and other regulatory authorities, expectant mothers we care for or third parties, that could have a material adverse effect on our business, consolidated financial condition, results of operations, cash flows and liquidity. Any such proceeding or action, any related indemnification obligation, even if we are not held liable, and any resulting negative publicity, could harm our business, damage our reputation, force us to incur significant expenses in defense of these proceedings, increase the costs of conducting our business, distract the attention of management or result in the imposition of financial liability.
26
We may be required to expend significant capital and other resources to protect against the threat of cybersecurity attacks and security breaches or to alleviate problems caused by breaches, including unauthorized access to data regarding expectant mothers and unborn babies and personally identifiable information stored in our information systems, the introduction of computer viruses or other malicious software programs to our systems, cybersecurity attacks, email phishing schemes, network disruption, denial of service attacks, malware and ransomware. A cybersecurity attack or other incident that bypasses our, the expectant mothers we care for or third-party service providers’ information system’s security could cause a security breach that may lead to a material disruption to our information systems infrastructure or business and may involve a significant loss of business or patient health information and other confidential, sensitive or personal information. If a cybersecurity attack or other unauthorized attempt to access our systems or facilities, or those of the expectant mothers we care for or third-party service providers, were to be successful, it could result in the theft, destruction, loss, misappropriation or release of confidential, sensitive or personal information or intellectual property, and could cause operational or business delays that may materially impact our ability to provide various services. Any successful cybersecurity attack or other unauthorized attempt to access our systems or facilities, or those of the expectant mothers we care for or third-party service providers, also could result in negative publicity which could damage our reputation or brand with the expectant mothers we care for, referral sources, payers or other third parties and could subject us to substantial sanctions, fines and damages and other additional civil and criminal penalties under the Health Insurance Portability and Accountability Act, or HIPAA, the Health Information Technology for Economic and Clinical Health Act, or HITECH Act, the HIPAA Omnibus Rule, and other federal and state privacy laws, in addition to litigation with those affected.
We and our third-party service providers may become the victims of these types of threats, attacks and security breaches. No security measures, procedures, technology or amount of preparation can provide guaranteed protection from these threats, or ensure that we, the expectant mothers we care for and our third-party service providers will not be victims in the future. Cybersecurity attacks may disrupt, or result in unauthorized access to, our networks, applications and confidential, personal or sensitive data, and those of the expectant mothers we care for or service providers, and successful attacks may occur in the future.
Our information systems require an ongoing commitment of significant resources to maintain, protect and enhance our existing systems. As we expand our business, we will need to continue to scale our information technology systems and personnel to support our growth, including the manufacture and supply chain management of our INVU platform. Difficulties with implementing new technology systems, delays in our timeline for planned improvements, significant system failures or our inability to successfully modify our information systems to respond to changes in our business needs may cause disruptions in our business operations and adversely affect our business, financial condition and results of operations. Failure to maintain the security and functionality of our information systems and related software, or to defend a cybersecurity attack or other attempt to gain unauthorized access to our systems, facilities or health information regarding expectant mothers and unborn babies could expose us to a number of adverse consequences, the vast majority of which are not insurable, including but not limited to disruptions in our operations, regulatory and other civil and criminal penalties, fines, investigations and enforcement actions (including, but not limited to, those arising from the SEC, Federal Trade Commission, the HHS Office of Inspector General, or OIG, or State Attorneys General), litigation with those affected by the data breach, loss of expectant mothers wanting to utilize our services, disputes with payers and increased operating expense, which either individually or in the aggregate could have a material adverse effect on our business, financial position, results of operations and liquidity.
Our collection, use, storage, disclosure, transfer and other processing of personal information, could give rise to significant costs, liabilities and other risks, including as a result of investigations, inquiries, litigation, fines, legislative and regulatory action and negative press about our privacy and data protection practices, which may harm our business, financial conditions, results of operations and prospects.
In the course of our operations, we collect, use, store, disclose, transfer and otherwise process an increasing volume of personal information, including from expectant mothers and their clinicians, customers, partners, candidates and employees, consultants, website visitors, leads and third parties with whom we conduct business. Additionally, our expansion plans for our INVU platform contemplate our collection, use and storage of an increasing amount of personal health data. The collection, use, storage, disclosure, transfer and other processing of personal information is increasingly subject to a wide array of federal, state and foreign laws and regulations regarding data privacy and security, including comprehensive laws of broad application, such as the EU General Data Protection Regulation (EU) 2016/679, or GDPR, which is applicable across all member states of the European Economic Area. The GDPR as transposed into the national laws of the UK (the “UK GDPR”), protects the privacy of personal information collected, used, stored, disclosed, transferred and otherwise processed in or from the governing jurisdiction. Our ability to collect, use, maintain or otherwise process personal data has been, and could be further, restricted by additional laws such as the California Consumer Privacy Act (the “CCPA”) and the Israeli Privacy Protection Law, 1981 and the regulations thereunder, or the Israeli Privacy Law. A current pending amendment to the Israeli Privacy Law, if passed into law, may enhance fines and sanctions for breaching the Israeli Privacy Law and to strengthen the enforcement capacity of the Israeli Privacy Protection Authority.
27
These laws and regulations generally define personal data to include location data and online identifiers, which are commonly used and collected parameters in digital advertising and, among other things, impose stringent user consent requirements and permit data subjects to request we discontinue using certain data. In addition, some countries are considering or have enacted legislation requiring local storage and processing of data that could increase the cost and complexity of delivering our services.
Additionally, the uncertainty created by these laws and regulations can be compounded when services hosted in one jurisdiction are directed at users in another jurisdiction. The GDPR has a wide territorial scope and contains significant penalties for non-compliance. The GDPR, among other things, imposes requirements to provide detailed and transparent disclosures about how personal data is collected and processed, grants rights for data subjects to access, delete or object to the processing of their personal data, provides for mandatory breach notification to supervisory authorities (and in certain cases, affected individuals) of certain data breaches, sets limitations on the retention of personal data and outlines significant documentary requirements to demonstrate compliance through policies, procedures, training and audits. Additionally, supervisory authorities in the member states have some flexibility when implementing European directives and certain aspects of the GDPR, which can lead to diverging national rules. European supervisory authorities have been very active in terms of enforcing data protection rules, including with respect to cookie-related matters. Regulation of cookies and similar technologies, and any decline of cookies or similar online tracking technologies as means to identify and potentially target individuals, may lead to broader restrictions and impairments on our business and, specifically, online activities.
As we seek to expand our business, we are, and may increasingly become, subject to various laws, regulations and standards, as well as contractual obligations, relating to data privacy and security in the jurisdictions in which we operate. When conducting clinical trials, we face risks associated with collecting trial participants’ data, especially health data, in a manner consistent with applicable laws and regulations, such as GCP guidelines or FDA human subject protection regulations. The GDPR/UK GDPR would also increase our obligations with respect to any clinical trials conducted in the EEA/UK, requiring changes to informed consent practices and more detailed notices for clinical trial subjects and investigators. In particular, the processing of ’special category data’ (such as personal data relating to health and genetic information), which will be relevant to our operations in the context of our conduct of clinical trials, imposes heightened compliance burdens under European and UK data protection laws.
In many cases, these laws and regulations apply not only to third-party transactions, but also to transfers of information between or among us, any affiliates and other parties with whom we conduct business. These laws, regulations and standards may be interpreted and applied differently over time and from jurisdiction to jurisdiction, and it is possible that they will be interpreted and applied in ways that may harm our business, financial condition and results of operations. The regulatory framework for data privacy and security worldwide is continuously evolving and developing and, as a result, interpretation and implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future.
We are subject to diverse laws and regulations relating to data privacy and security. The GDPR/UK GDPR is wide-ranging in scope and imposes numerous, significant and complex requirements on organizations that process personal data, including (without limitation) requirements relating to processing health and other sensitive data, establishing a legal basis for any processing of personal data, obtaining consent of the individuals to whom the personal data relates, providing information to individuals regarding data processing activities, limiting the collection and retention of personal data through ‘data minimization’ and ’storage limitation’ principles, implementing safeguards to protect the security and confidentiality of personal data, honoring increased rights for data subjects, providing notification of data breaches in some instances and taking certain measures when engaging third-party processors.
28
In the United States, various federal and state regulators have adopted, or are considering adopting, laws and regulations concerning personal information and data security, including HIPAA. This patchwork of legislation and regulation may give rise to conflicts or differing views of personal privacy rights. For example, certain state laws may be more stringent or broader in scope, or offer greater individual rights, with respect to personal information than federal, international or other state laws, and such laws may differ from each other, all of which may complicate compliance efforts. Additionally, new privacy rules are being enacted in the United States and globally, and existing ones are being updated and strengthened. For example, the CCPA, which increases privacy rights for California residents and imposes obligations on companies that process their personal information, came into effect on January 1, 2020. Among other things, the CCPA requires covered companies to provide new disclosures to California consumers and provide such consumers new data protection and privacy rights, including the ability to opt-out of certain sales of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for certain data breaches that result in the loss of personal information. This private right of action may increase the likelihood of, and risks associated with, data breach litigation. The CCPA was amended in September 2018 and November 2019, and on November 3, 2020, California voters approved a new privacy law, the California Privacy Rights Act, or the CPRA, which significantly modifies the CCPA, including by expanding consumers’ rights with respect to certain personal information and creating a new state agency to oversee implementation and enforcement efforts. Many of the CPRA’s provisions became effective on January 1, 2023. It is possible that further amendments will be enacted, but even in its current form it remains unclear how various provisions of the CCPA will be interpreted and enforced. State laws are changing rapidly and there is discussion in Congress of a new federal data protection and privacy law to which we would become subject if it is enacted. All of these evolving compliance and operational requirements impose significant costs that are likely to increase over time, may require us to modify our data processing practices and policies, divert resources from other initiatives and projects, and could restrict the way products and services involving data are offered, all of which may harm our business, financial condition and results of operations.
The GDPR/UK GDPR also imposes strict rules on the transfer of EEA/UK personal data to countries outside the EEA/UK. The GDPR/UK GDPR generally prohibits the transfer of EEA and UK personal data to third countries whose laws do not ensure an adequate level of protection, unless a valid data transfer mechanism has been implemented or an Article 49 GDPR/UK GDPR derogation applies. Recent legal developments in the EEA and UK have created complexity and uncertainty regarding transfers of personal data. On 16 July 2020, the Court of Justice of the European Union issued its judgement in Schrems II, which invalidated the EU-US Privacy Shield as a valid data transfer mechanism. The decision upheld the use of the European Commission Standard Contractual Clauses, or SCCs, as a valid data transfer mechanism, but required organizations to take supplementary measures when relying on the SCCs. As supervisory authorities issue further guidance on personal data export mechanisms, including circumstances where the SCCs cannot be used, and/or start taking enforcement action, we could suffer additional costs, complaints and/or regulatory investigations or fines, and/or if we are otherwise unable to transfer personal data between and among countries and regions in which we operate, it could affect the manner in which we operate our business and could harm our business, financial condition and results of operations. On June 4, 2021, the European Commission published a new set of modular SCCs. The new SCCs also apply only to the transfer of data outside of the EEA and not the UK. Although the European Commission adopted an adequacy decision for the UK on 28 June 2021, allowing the continued flow of personal data from the EEA to the UK, this decision will be regularly reviewed going forward and may be revoked if the UK diverges from its current adequate data protection laws following its exit from the European Union. In addition, the UK Information Commissioner’s Office, or ICO, has undergone a period of public consultation on its own specific international data transfer agreement. We are monitoring these developments, but we may, in addition to other impacts, experience additional costs associated with increased compliance burdens and be required to engage in new contract negotiations with third parties that aid in processing data on our behalf or localize certain data. We may also experience reluctance or refusal by prospective European customers to use our solutions, and we may find it necessary or desirable to make further changes to our handling of personal data of EEA- and UK-based data subjects. In addition, transfers of personal data outside of the UK are subject to the UK GDPR which restricts and limits our ability to transfer personal data globally.
Effective as of July 17, 2023, the EU-U.S. Data Privacy Framework (EU-U.S. DPF), the UK Extension to the EU-U.S. Data Privacy Framework (UK Extension to the EU-U.S. DPF), and the Swiss-U.S. Data Privacy Framework (Swiss-U.S. DPF) were respectively developed in furtherance of transatlantic commerce by the U.S. Department of Commerce and the European Commission, the UK Government, and the Swiss Federal Administration to provide U.S. organizations with reliable mechanisms for personal data transfers to the United States from the European Union European Economic Area, the United Kingdom (and Gibraltar), and Switzerland.
29
While the DPF meant to enable an easier path for transfers of personal data to the U.S., it includes numerous obligation and complexities and therefore creates liabilities, costs and legal exposure to a U.S. entity that chose to use it as transfer method. Traditionally and repeatedly mechanisms for transferring information to the U.S. have been rejected by the Court of Justice of the European Union and therefore uncertainty surrounds the issue, and it is unclear whether the DPF will last and for how long. This causes uncertainty and may lead to legal and technological expenses.
In addition to government regulation, privacy advocates and industry groups have and may in the future propose self-regulatory standards from time to time. These and other industry standards may legally or contractually apply to us, or we may elect to comply with such standards. We expect that there will continue to be new proposed laws and regulations concerning data privacy and security, and we cannot yet determine the impact such future laws, regulations and standards may have on our business. New laws, amendments to or re-interpretations of existing laws, regulations, standards and other obligations may require us to incur additional costs and restrict our business operations. Because the interpretation and application of laws, regulations, standards and other obligations relating to data privacy and security are still uncertain, it is possible that these laws, regulations, standards and other obligations may be interpreted and applied in a manner that is inconsistent with our data processing practices and policies or the features of our INVU platform and services. If so, in addition to the possibility of fines, lawsuits, regulatory investigations, public censure, other claims and penalties, and significant costs for remediation and damage to our reputation, we could be materially and adversely affected if legislation or regulations are expanded to require changes in our data processing practices and policies or if governing jurisdictions interpret or implement their legislation or regulations in ways that negatively impact our business, financial condition and results of operations. We may be unable to make such changes and modifications in a commercially reasonable manner, or at all. In relation to enforcement under the GDPR/UK GDPR, European and UK data protection laws now also provide for greater penalties for non-compliance than previous data protection laws, including, for example, separate administrative fines ranging from €10 million/£8.7 million to €20 million/£17.5 million or 2% to 4% of global annual revenue of any non-compliant organization for the preceding financial year, whichever is higher. Any inability to adequately address data privacy or security-related concerns, even if unfounded, or to comply with applicable laws, regulations, standards and other obligations relating to data privacy and security, could result in additional cost and liability to us, harm our reputation and brand, damage our relationships with consumers and harm our business, financial condition and results of operations.
We make public statements about our use and disclosure of personal information through our privacy policies, information provided on our website and press statements. Although we endeavor to comply with our public statements and documentation, we may at times fail to do so or be alleged to have failed to do so. The publication of our privacy policies and other statements that provide promises and assurances about data privacy and security can subject us to potential government or legal action if they are found to be deceptive, unfair or misrepresentative of our actual practices. Furthermore, in the EEA/UK, regulators are increasingly focusing on compliance with requirements in the online behavioral advertising ecosystem, and current national laws that implement the ePrivacy Directive are highly likely to be replaced in Europe by a regulation known as the ePrivacy Regulation, which will significantly increase fines for non-compliance, noting that the ePrivacy Regulation when it comes into effect will have no bearings on the UK in a post-Brexit world. Recent guidance and case law in the European Union and UK require opt-in, informed consent for the placement of a cookie or similar tracking technologies on a customer’s device and for direct electronic marketing. The GDPR/UK GDPR also imposes conditions on obtaining valid consent, such as a prohibition on pre-checked consents and a requirement to ensure separate consents are sought for each type of cookie or tracking technology. While the text of the ePrivacy Regulation is still under development, recent European case law and regulators’ recent guidance are driving increased attention to cookies and tracking technologies. This could lead to substantial costs, require significant systems changes, limit the effectiveness of our marketing activities, divert the attention of our technology personnel, adversely affect our margins, increase costs and subject us to additional liabilities. Regulation of cookies and similar technologies, and any decline of cookies or similar online tracking technologies as a means to identify and potentially target users, may lead to broader restrictions and impairments on our marketing and personalization activities and may negatively impact our efforts to understand our customers. Any concerns about our data privacy and security practices, even if unfounded, could damage the reputation of our business and harm our business, financial condition and results of operations.
30
Complying with these numerous, complex and often changing regulations is expensive and difficult. Any failure or perceived failure by us or our service providers to comply with our posted privacy policies or with any applicable federal, state or similar foreign laws, regulations, standards, certifications or orders relating to data privacy, security or consumer protection, or any compromise of security that results in the theft, unauthorized access, acquisition, use, disclosure or misappropriation of personal information or other user data, could result in significant fines or penalties, negative publicity or proceedings or litigation by governmental agencies or consumers, including class action privacy litigation in certain jurisdictions, which would subject us to significant awards, penalties or judgments, one or all of which could require us to change our business practices or increase our costs and could materially and adversely affect our business, financial condition and results of operations. In addition, if our practices are not consistent, or viewed as not consistent, with legal and regulatory requirements, including changes in laws, regulations and standards or new interpretations or applications of existing laws, regulations and standards, we may also become subject to audits, inquiries, whistleblower complaints, adverse media coverage, investigations, criminal or civil sanctions, all of which may harm our business, financial condition and results of operations.
Navigating the rapidly changing landscape of data protection and privacy regulations is both challenging and costly. Failure to comply with such regulations, whether in our practices or those of our service providers, can lead to substantial fines, legal action, and negative publicity. As the regulatory environment constantly evolves, especially with emerging technologies like artificial intelligence and machine learning, adhering to these laws can restrict our operations, expose us to legal risks, and take compliance costs. Additionally, inconsistencies with legal requirements may trigger audits, investigations, and sanctions, all of which could significantly impact our business, finances, and operations.
We could become subject to product liability claims, product recalls, and warranty claims that could be expensive, divert management’s attention and harm our business reputation and financial results.
Our business exposes us to potential liability risks that are inherent in the marketing and sale of products used in healthcare. We may be held liable if our INVU platform or if any other product that integrates our technology causes injury or death or is found otherwise unsafe or unsuitable during usage, including misuse by the user or by care providers, whether or not such use is consistent with our products’ instructions. Additionally, while our INVU platform is currently cleared to measure FHR, MHR and MUA during the antepartum period, and as a result, offer NSTs, we plan to significantly expand our INVU platform’s permitted uses; and the added complexity may expose us to additional potential liability, including if our INVU platform provides incorrect data that leads to missed complications, false positives or negatives or other reported results that are inconsistent with otherwise accurate readings. Our INVU platform incorporates sophisticated components and computer software. Complex software can contain errors, particularly when first introduced. In addition, new products or enhancements may contain undetected errors or performance problems that, despite testing, are discovered only after installation. Patients could allege or possibly prove defects of our INVU platform or other products that integrate our technology. Additionally, disruptions in access to or availability of the cloud-based services on which our INVU platform will rely, whether due to service interruptions, cyberattacks or other reasons, could result in product liability issues, including as a result of the failure of our INVU platform to timely provide results to healthcare professionals.
A product liability claim, regardless of its merit or eventual outcome, could result in significant legal defense costs and divert management’s attention. Regardless of merit or eventual outcome, liability claims may result in:
| ● | decreased demand for our INVU platform; |
| ● | injury to our reputation; |
| ● | costs of related litigation; |
| ● | substantial monetary awards to patients and others; |
| ● | loss of revenue; and |
| ● | the inability to commercialize future products. |
31
Any of these outcomes may have an adverse effect on our business, financial condition and results of operations, and may increase the volatility of our share price.
The coverage limits of our insurance policies we may choose to purchase to cover related risks may not be sufficient to cover future claims. If sales of our INVU platform or other products integrating our technology increase or we suffer future product liability claims, we may be unable to maintain product liability insurance at satisfactory rates or with adequate amounts or at all. A product liability claim, any product recalls or excessive warranty claims, whether arising from defects in design or manufacture or otherwise, could negatively affect our sales or require a change in the design or manufacturing process, any of which could harm our relationship with our customers and partners, and have a material adverse impact on our reputation and business, financial condition, results of operations and prospects.
In addition, if our INVU platform or other products integrating our technology are defective, we, our future customers or partners may be required to notify regulatory authorities and/or to recall the products. Any recall would divert management’s attention and financial resources and harm our reputation with customers, patients, medical professionals and third-party payers. A recall involving our INVU platform would be particularly harmful to our business. The adverse publicity resulting from any of these actions could adversely affect the perception of our customers or partners. These investigations or recalls, especially if accompanied by unfavorable publicity, could result in our incurring substantial costs, losing revenues and damaging our reputation, each of which would harm our business, financial condition, results of operations and prospects.
Our INVU platform is not yet approved for third-party payer coverage or reimbursement. If in the future we are approved for and are otherwise able to commercialize it, but are unable to obtain adequate reimbursement or insurance coverage from third-party payers, we may not be able to generate significant revenue, in which case we may need to obtain additional financing.
Our INVU platform is not yet approved for third-party payer coverage or reimbursement. Coding and coverage determinations as well as reimbursement levels and conditions are important to the commercial success of our INVU platform. The future availability of insurance coverage and reimbursement for newly approved medical devices is highly uncertain, and our future business will be greatly impacted by the level of reimbursement provided by third-party payers. In the United States, third-party payers decide which products and services they will cover, how much they will pay and whether they will continue reimbursement. Third-party payers may not cover or provide adequate reimbursement for our INVU platform or the related services, assuming we are able to fully develop and obtain all regulatory approvals and clearances to market it in the United States or other geographies. Accordingly, unless government and other third-party payers provide coverage and reimbursement for our services, patients and healthcare providers may choose not to use them, which would cause investors to lose their entire investment. A primary trend in the United States healthcare industry and elsewhere is cost containment. Government authorities and other third-party payers have attempted to control costs by limiting coverage and the amount of reimbursement for particular products and services. Reimbursement may not be available, or continue to be available, for our INVU platform, other products or systems using our technology or any other products we may develop in the future, or even if reimbursement is available, such reimbursement may not be adequate. We also will be subject to foreign reimbursement policies in the international markets we expect to enter. Decisions by health insurers or other third-party payers in these markets not to cover, or to discontinue reimbursing, our INVU platform could materially and adversely affect our business. If such decisions are made, they could also have a negative impact on our ability to generate revenues, in which case we may need to obtain additional financing.
Any default under Nuvo’s Bridge Financing Notes could have significant consequences.
The Bridge Financing Notes are secured by Nuvo’s intellectual property, evidenced by collateral assignments/financing statements Nuvo has filed with the United States Patent & Trademark Offices and is in the process of filing with Nuvo’s Registrar in Israel. The Bridge Financing Notes contain certain covenants, including regarding the payment of interest thereon, use of proceeds, and conduct of business, as well as representations and warranties made to the holders of such notes.
Nuvo’s ability to comply with these covenants in the Bridge Financing Notes may be affected by events beyond its control, including prevailing economic, financial and industry conditions. The breach of any of these covenants could result in an event of default, which would permit the holders of the Bridge Financing Notes to declare all outstanding debt to be due and payable, together with accrued and unpaid interest. If Nuvo is unable to repay the accelerated amounts, the holders of the Bridge Financing Notes could proceed against the collateral granted to them to secure such debt. If the payment of Bridge Financing Notes is accelerated, Nuvo’s assets may be insufficient to repay such debt in full, which could result in its insolvency. Any default by Nuvo under the Bridge Financing Notes could have a material adverse effect on its business, financial condition and results of operations.
32
Risks Related to Government Regulation and Our Industry
We conduct business in a heavily regulated industry, and changes in regulations or violations of regulations may, directly or indirectly, reduce our revenue, adversely affect our results of operations and financial condition and harm our business.
The health care industry is highly regulated, and the regulatory environment in which we operate may change significantly and adversely to us in the future. Areas of the regulatory environment that may affect our ability to conduct business include, without limitation:
| ● | federal and state laws applicable to medical device ordering, documentation of medical devices ordered, billing practices and claims payment and/or regulatory agencies enforcing those laws and regulations, including state licensing laws; |
| ● | federal and state fraud and abuse laws; |
| ● | federal and state laws applicable to pre-clinical and clinical human subject trials; |
| ● | coverage and reimbursement levels by Medicare, Medicaid, other governmental payers and private insurers; |
| ● | restrictions on coverage of, and reimbursement for, medical devices; |
| ● | federal and state Occupational Safety and Health Administration rules and regulations; and |
| ● | HIPAA, and similar state data privacy laws. |
If we fail to comply with U.S. federal and state fraud and abuse and other healthcare laws and regulations, including those relating to kickbacks and false claims for reimbursement, we could face substantial penalties and our business operations and financial condition could be harmed.
Healthcare providers play a primary role in the distribution, recommendation, ordering and purchasing of any medical device for which we have or obtain marketing clearance or approval. Through our arrangements with healthcare professionals and customers, we are exposed to broadly applicable anti-fraud and abuse, anti-kickback, false claims and other healthcare laws and regulations that may constrain our business, our arrangements and relationships with customers, and how we market, sell and distribute our marketed medical device. We have a compliance program, code of conduct and associated policies and procedures, but it is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent noncompliance may not be effective in protecting us from governmental investigations for failure to comply with applicable fraud and abuse or other healthcare laws and regulations.
In the United States, we are subject to various state and federal anti-fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and federal False Claims Act, or the FCA. There are similar laws in other countries. Our relationships with physicians, other health care professionals and hospitals, and obstetrician physician practice management groups are subject to scrutiny under these laws.
The laws that may affect our ability to operate include, among others:
| ● | The federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willingly soliciting, offering, receiving or paying remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual, or the purchase, order or recommendation of, items or services for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value, and the government can establish a violation of the Anti-Kickback Statute without proving that a person or entity had actual knowledge of the law or a specific intent to violate. In addition, the government may assert that a claim, including items or services resulting from a violation of the Anti-Kickback Statute, constitutes a false or fraudulent claim for purposes of the FCA. There are a number of statutory exceptions and regulatory safe harbors protecting certain business arrangements from prosecution under the Anti-Kickback Statute; however, those exceptions and safe harbors are drawn narrowly, and there may be limited or no exception or safe harbor for many common business activities. Certain common business activities including, certain reimbursement support programs, educational and research grants or charitable donations, and practices that involve remuneration to those who prescribe, purchase or recommend medical devices, including discounts, providing items or services for free or engaging such individuals as consultants, advisors or speakers, may be subject to scrutiny if they do not fit squarely within an exception or safe harbor and would be subject to a facts and circumstances analysis to determine compliance with the Anti-Kickback Statute. Our business may not in all cases meet all of the criteria for statutory exception or regulatory safe harbor protection from anti-kickback liability; |
33
| ● | Federal, civil and criminal false claims laws, including the FCA, and civil monetary penalties laws, which prohibit, among other things, persons or entities from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds and knowingly making, using or causing to be made or used, a false record or statement to get a false claim paid or to avoid, decrease or conceal an obligation to pay money to the federal government. A claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. Actions under the FCA may be brought by the government or as a qui tam action by a private individual in the name of the government. These individuals, sometimes known as “relators” or, more commonly, as “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. Many medical device manufacturers have been investigated and have reached substantial financial settlements with the federal government under the FCA for a variety of alleged improper activities, including causing false claims to be submitted as a result of the marketing of their products for unapproved and thus non-reimbursable uses and interactions with prescribers and other customers, including those that may have affected their billing or coding practices and submission of claims to the federal government. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and mandatory monetary penalties for each false or fraudulent claim or statement. Because of the potential for large monetary exposure, healthcare and medical device companies often resolve allegations without admissions of liability for significant and material amounts to avoid the uncertainty of treble damages and per claim penalties that may be awarded in litigation proceedings; |
| ● | HIPAA, which imposes criminal and civil liability for, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers, or knowingly and willfully falsifying, concealing or covering up a material fact or making a materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services; |
| ● | HIPAA, as amended by the HITECH Act, and their implementing regulations, also impose obligations, including mandatory contractual terms, on covered entities subject to the rule, such as health plans, healthcare clearinghouses and certain healthcare providers, as well as their business associates and their covered subcontractors that perform certain services for them or on their behalf involving the use or disclosure of individually identifiable health information with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| ● | The federal Physician Payments Sunshine Act, also known as Open Payments, which requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually, with certain exceptions to the Centers for Medicare & Medicaid Services, or CMS, information related to payments or other “transfers of value” made to physicians, defined to include doctors, dentists, optometrists, podiatrists and chiropractors, and teaching hospitals, and requires applicable manufacturers and group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding payments and transfers of value provided, as well as ownership and investment interests held, during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified nurse anesthetists and certified nurse-midwives; and |
| ● | Analogous state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers; state laws that require medical device companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state beneficiary inducement laws, which are state laws that require medical device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
34
State and federal regulatory and enforcement agencies continue to actively investigate violations of healthcare laws and regulations, and the U.S. Congress continues to strengthen the arsenal of enforcement tools. Most recently, the Bipartisan Budget Act of 2018, or the BBA, increased the criminal and civil penalties that can be imposed for violating certain federal health care laws, including the Anti-Kickback Statute. Enforcement agencies also continue to pursue novel theories of liability under these laws. In particular, government agencies have increased regulatory scrutiny and enforcement activity with respect to manufacturer reimbursement support activities and patient support programs, including bringing criminal charges or civil enforcement actions under the Anti-Kickback Statute, federal FCA and HIPAA’s healthcare fraud and privacy provisions.
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available under such laws, it is possible that some of our business activities, including certain sales and marketing practices of our INVU platform, and financial arrangements with physicians, other healthcare providers, and other customers, could be subject to challenge under one or more such laws. If an arrangement were deemed to violate the Anti-Kickback Statute, it may also subject us to violations under other fraud and abuse laws such as the federal FCA and civil monetary penalties laws. Moreover, such arrangements could be found to violate comparable state fraud and abuse laws.
Achieving and sustaining compliance with applicable federal and state anti-fraud and abuse laws may prove costly. If we, our employees or our contractors are found to have violated any of the above laws we may be subjected to substantial criminal, civil and administrative penalties, including imprisonment, exclusion from participation in federal healthcare programs, such as Medicare and Medicaid, and significant fines, monetary penalties, forfeiture, disgorgement and damages, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results. Any action or investigation against us for the violation of these healthcare fraud and abuse laws, even if successfully defended, could result in significant legal expenses and could divert our management’s attention from the operation of our business. Companies settling federal FCA, Anti-Kickback Statute or civil monetary penalties law cases also may be required to enter into a Corporate Integrity Agreement with the OIG, in order to avoid exclusion from participation (such as loss of coverage for their products) in federal healthcare programs such as Medicare and Medicaid. Corporate Integrity Agreements typically impose substantial costs on companies to ensure compliance. Defending against any such actions can be costly, time-consuming and may require significant personnel resources, and may harm our business, financial condition and results of operations.
In addition, the medical device industry’s relationship with physicians is under increasing scrutiny by the OIG, the U.S. Department of Justice, or the DOJ, the state attorney generals and other foreign and domestic government agencies. Our failure to comply with requirements governing the industry’s relationships with physicians or an investigation into our compliance by the OIG, the DOJ, state attorney generals and other government agencies, could harm our business, financial condition and results of operations.
Our employees, independent contractors, consultants, commercial partners and suppliers may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could harm our business, financial condition and results of operations.
We are exposed to the risk that our employees, independent contractors, consultants, commercial partners, suppliers and distributors may engage in fraudulent or illegal activity. Misconduct by these parties could include intentional, reckless or negligent conduct or disclosure of unauthorized activities to us that violates: (i) the rules and regulations of the FDA and other similar foreign regulatory bodies, including those laws requiring the reporting of true, complete and accurate information to such regulators; (ii) manufacturing standards; (iii) healthcare fraud and abuse laws in the United States and similar foreign fraudulent misconduct laws; or (iv) laws that require the true, complete and accurate reporting of financial information or data. These laws may impact, among other things, future sales, marketing and education programs. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry, are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commissions, certain customer incentive programs and other business arrangements generally. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials.
35
We have adopted a code of conduct, but it is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent these activities may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could result in the imposition of significant fines or other sanctions, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, imprisonment, additional integrity reporting and oversight obligations, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings and curtailment of operations, any of which could adversely affect our ability to operate our business and our results of operations. Whether or not we are successful in defending against any such actions or investigations, we could incur substantial costs, including legal fees, and divert the attention of management in defending ourselves against any of these claims or investigations, which could harm our business, financial condition and results of operations.
Regulatory compliance is expensive, complex and uncertain, and a failure to comply could lead to enforcement actions against us and other negative consequences for our business.
The FDA and similar agencies regulate our INVU platform as a medical device. Complying with these regulations is costly, time-consuming, complex and uncertain. For instance, before a new medical device, or a new intended use for, an existing device can be marketed in the United States, a company must first submit and receive either a 510(k) clearance, de novo authorization or approval of a PMA from the FDA, unless an exemption applies. FDA regulations and regulations of similar agencies are wide-ranging and include, among other things, oversight of:
| ● | product design, development, manufacturing (including suppliers) and testing; |
| ● | laboratory, preclinical and clinical studies; |
| ● | product safety and effectiveness; |
| ● | product labeling; |
| ● | product storage and shipping; |
| ● | record keeping; |
| ● | pre-market clearance or approval; |
| ● | marketing, advertising and promotion; |
| ● | product sales and distribution; |
| ● | product changes; |
| ● | product recalls; and |
| ● | post-market surveillance and reporting of deaths or serious injuries and certain malfunctions. |
Our INVU platform is subject to extensive regulation by the FDA and non-U.S. regulatory agencies. Further, improvements of our INVU platform and any potential new products will be subject to extensive regulation and will likely require permission from regulatory agencies and ethics boards to conduct clinical trials and clearance or approval from the FDA and non-U.S. regulatory agencies prior to commercial sale and distribution. Failure to comply with applicable U.S. requirements regarding, for example, promoting, manufacturing or labeling our INVU platform may subject us to a variety of administrative or judicial actions and sanctions, such as Form 483 observations, warning letters, untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution. Any enforcement action by the FDA and other comparable non-U.S. regulatory agencies could harm our business, financial condition and results of operations.
36
Our failure to comply with applicable regulatory requirements could result in enforcement action by the FDA or state agencies, which may include any of the following actions:
| ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| ● | unanticipated expenditures to address or defend such actions; |
| ● | customer notifications for repair, replacement or refunds; |
| ● | recall, detention or seizure of our INVU platform; |
| ● | operating restrictions or partial suspension or total shutdown or production; |
| ● | refusing or delaying our requests for 510(k) clearance or PMA of new products or modified products; |
| ● | operating restrictions; |
| ● | withdrawing 510(k) clearances or PMAs that have already been granted; |
| ● | refusal to grant export approval for our INVU platform; or |
| ● | criminal prosecution. |
If any of these events were to occur, it would have a negative impact on our business, financial condition and results of operations.
The FDA also regulates the advertising and promotion of our INVU platform to ensure that the claims we make are consistent with our regulatory clearances and approvals, that there are adequate and reasonable data to substantiate the claims and that our promotional labeling and advertising is neither false nor misleading in any respect. If the FDA determines that any of our advertising or promotional claims are misleading, not substantiated or not permissible, we may be subject to enforcement actions, including warning letters, and we may be required to revise our promotional claims and make other corrections or restitutions.
Our medical device operations are subject to pervasive and continuing FDA regulatory requirements, and failure to comply with these requirements could harm our business, financial condition and results of operations.
Medical devices regulated by the FDA are subject to “general controls” which include: registration with the FDA; listing commercially distributed products with the FDA; complying with GMPs under QSR; filing reports with the FDA of, and keeping records relative to certain types of adverse events associated with devices under the medical device reporting regulation; assuring that device labeling complies with device labeling requirements; reporting certain device field removals and corrections to the FDA; and obtaining pre-market notification 510(k) clearance for devices prior to marketing. Some devices known as “510(k)-exempt” devices can be marketed without prior marketing-clearance or approval from the FDA. The current indications for use and the passive nature of our platform and hardware classifies our product as a Class II product, although future services or tools may subject our product to different classifications or regulatory pathways.
The medical device industry is now experiencing greater scrutiny and regulation by federal, state and foreign governmental authorities. Companies in our industry are subject to more frequent and more intensive reviews and investigations, often involving the marketing, business practices and product quality management. Such reviews and investigations may result in civil and criminal proceedings; the imposition of substantial fines and penalties; the receipt of warning letters, untitled letters, demands for recalls or the seizure of our INVU platform; the requirement to enter into corporate integrity agreements, stipulated judgments or other administrative remedies; and result in our incurring substantial unanticipated costs and the diversion of key personnel and management’s attention from their regular duties, any of which may harm our business, financial condition and results of operations, and may result in greater and continuing governmental scrutiny of our business in the future.
37
Additionally, federal, state and foreign governments and entities have enacted laws and issued regulations and other standards requiring increased visibility and transparency of our interactions with healthcare providers. For example, Open Payments requires us to annually report to CMS payments and other transfers of value to all U.S. physicians and U.S. teaching hospitals, with the reported information made publicly available on a searchable website. Failure to comply with these legal and regulatory requirements could impact our business, and we have had and will continue to spend substantial time and financial resources to develop and implement enhanced structures, policies, systems and processes to comply with these legal and regulatory requirements, which could harm our business, financial condition and results of operations.
Material modifications to our INVU platform may require new 510(k) clearances or pre-market approvals or may require us to recall or cease marketing our INVU platform until clearances or approvals are obtained, which could harm our business, financial condition and results of operations.
Modifications that could significantly affect the safety and effectiveness of our approved or cleared products, such as changes to the intended use or technological characteristics of our INVU platform, will require new 510(k) clearances or PMAs or require us to recall or cease marketing the modified device until these clearances or approvals are obtained. Based on FDA published guidelines, the FDA requires device manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplemental approval or clearance; however, the FDA can review a manufacturer’s decision. Any modification to an FDA-cleared device that could significantly affect its safety or efficacy or that would constitute a major change in its intended use may require a new 510(k) clearance or possibly a PMA. We may not be able to obtain additional 510(k) clearances or PMAs for new products or for modifications to, or additional indications for, our INVU platform in a timely fashion, or at all. Delays in obtaining required future clearances or approvals could adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth. For example, the FDA may request additional or new clinical studies to prove safety or efficacy. We may make additional modifications to our INVU platform in the future which could require additional clearances or approvals. If the FDA requires new clearances or approvals for these modifications, we may be required to recall and to stop selling or marketing such product as modified, which could harm our operating results and require us to redesign such product. In these circumstances, we may be subject to significant enforcement actions. The FDA may also change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may impact our ability to modify our currently approved or cleared products on a timely basis. Any of these actions could harm our business, financial condition and results of operations.
There is no guarantee that the FDA will approve a de novo classification application, grant 510(k) clearance or pre-market approval of any material modifications to our INVU platform or for future products and failure to obtain necessary clearances or approvals would adversely affect our ability to grow our business.
In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, in order to clear the proposed device for marketing. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data is sometimes required to support substantial equivalence. In the de novo classification process, the FDA may classify a novel medical device as Class I or Class II if no predicate device is legally on the market.
The FDA’s de novo classification process generally takes six months from submission, but may take longer. The FDA’s 510(k) clearance process usually takes between three to 12 months from submission, but may last longer. The process of obtaining PMA approval is much more rigorous, costly, lengthy and uncertain than the 510(k) clearance process. It generally takes one to three years, or even longer, from the time the PMA is submitted to the FDA until an approval is obtained. In the PMA approval process, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data. Any delay or failure to obtain necessary regulatory approvals or clearances would have a material adverse effect on our business, financial condition and prospects.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| ● | our inability to demonstrate to the satisfaction of the FDA that our INVU platform is safe or effective for its intended uses; |
38
| ● | the FDA may request additional information following the receipt of a de novo classification application; |
| ● | the FDA may reject a de novo application upon identification of a legally marketed predicate device and require submission of a 510(k) clearance; |
| ● | our inability to establish substantial equivalence with a predicate device; |
| ● | the disagreement of the FDA with the design, conduct or implementation of our clinical trials or the analysis or interpretation of data from pre-clinical studies or clinical trials; |
| ● | serious and unexpected adverse device effects experienced by participants in our clinical trials; |
| ● | the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; |
| ● | our inability to demonstrate that the clinical and other benefits of the device outweigh the risks; |
| ● | an advisory committee, if convened by the FDA, may recommend against approval of our PMA or other application or may recommend that the FDA require, as a condition of approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions, or even if an advisory committee, if convened, makes a favorable recommendation, the FDA may still not approve the product; |
| ● | the FDA may identify deficiencies in our marketing application, and in our manufacturing processes, facilities or analytical methods or those of our third-party contract manufacturers; |
| ● | the potential for approval policies or regulations of the FDA or applicable foreign regulatory bodies to change significantly in a manner rendering our clinical data or regulatory filings insufficient for clearance or approval; and |
| ● | the FDA or foreign regulatory authorities may audit our clinical trial data and conclude that the data is not sufficiently reliable to support a PMA application. |
If we are unable to obtain approval for any medical device for which we plan to seek approval, our business may be harmed.
Although we have obtained regulatory clearance in the United States through MHR and FHR physiological measurements, as well as measuring MUA, and as a result, offering NSTs, and we have similarly obtained certification from the Medical Device Division, or AMAR, of the Ministry of Health in Israel, they will remain subject to extensive regulatory scrutiny. If regulatory sanctions are applied or if regulatory clearance or approval is withdrawn, it would harm our business, financial condition and results of operations.
Although we have obtained regulatory clearance in the United States through MHR and FHR physiological measurements, as well as measuring MUA, and as a result, offering NSTs, and we have similarly obtained certification from the AMAR in Israel, our INVU platform will be subject to ongoing regulatory requirements for manufacturing, distributing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies and submission of safety, effectiveness and other post-market information, including both federal and state requirements in the United States and requirements of comparable non-U.S. regulatory authorities.
Our manufacturing facilities are required to comply with extensive requirements imposed by the FDA and comparable foreign regulatory authorities, including ensuring that quality control and manufacturing procedures conform to the QSR or similar regulations set by foreign regulatory authorities. As such, we will be subject to continual review and inspections to assess compliance with the QSR and adherence to commitments made in any 510(k), de novo classification, or PMA application. Accordingly, we continue to spend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control.
39
Any regulatory clearances or approvals that we have received for our INVU platform will be subject to limitations on the cleared or approved indicated uses for which the product may be marketed and promoted, will be subject to the conditions of approval, or will contain requirements for potentially costly post-marketing testing. We are required to report certain adverse events and production problems, if any, to the FDA and comparable foreign regulatory authorities. Any new legislation addressing product safety issues could result in increased costs to ensure compliance. The FDA and other agencies, including the DOJ, closely regulate and monitor the post-clearance or approval marketing and promotion of products to ensure that they are marketed and distributed only for the cleared or approved indications and in accordance with the provisions of the cleared or approved labeling. We have to comply with requirements concerning advertising and promotion for our INVU platform.
Promotional communications with respect to devices are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the products’ cleared or approved labeling. As such, we may not promote our products for indications or uses for which they do not have clearance or approval. For certain changes to a cleared or approved product, including certain changes to product labeling, the holder of a cleared 510(k), de novo classification or approved PMA application may be required to submit a new application and obtain clearance or approval. We train our marketing and sales force against promoting our products for uses outside of the cleared or approved indications for use, known as off-label uses. However, physicians or healthcare providers may use our products for off-label purposes and are allowed to do so when in the physician’s independent professional medical judgment he or she deems it appropriate. If the FDA determines that our promotional materials or training constitute promotion of an off-label or other improper use, or that our internal policies and procedures are inadequate to prevent such off-label uses, it could subject us to regulatory or enforcement actions as discussed below.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with our facilities where the product is manufactured or disagrees with the promotion, marketing or labeling of a product, such regulatory agency may impose restrictions on that product or on us, including requiring withdrawal of the product from the market. If we fail to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may, among other things:
| ● | subject our facilities to an adverse inspectional finding or Form 483, or other compliance or enforcement notice, communication or correspondence; |
| ● | issue warning or untitled letters that would result in adverse publicity or may require corrective advertising; |
| ● | impose civil or criminal penalties; |
| ● | suspend or withdraw regulatory clearances or approvals; |
| ● | refuse to clear or approve pending applications or supplements to approved applications submitted by us; |
| ● | impose restrictions on our operations, including closing our suppliers’ facilities; |
| ● | seize or detain products; or |
| ● | require a product recall. |
In addition, violations of the Federal Food, Drug and Cosmetic Act, relating to the promotion of approved products may lead to investigations alleging violations of federal and state healthcare fraud and abuse and other laws, as well as state consumer protection laws.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and negatively impact our ability to commercialize and generate revenue from our INVU platform. If regulatory sanctions are applied or if regulatory clearance or approval is withdrawn, it would harm our business, financial condition and results of operations.
40
If we or our suppliers fail to comply with the FDA’s QSR, or any applicable state equivalent, our operations could be interrupted and our potential product sales and operating results could suffer.
Our manufacturing processes and those of our third-party suppliers are required to comply with the FDA’s QSR, which covers the design controls, document controls, purchasing controls, identification and traceability, production and process controls, acceptance activities, nonconforming product requirements, corrective and preventive action requirements, labeling and packaging controls, handling, storage, distribution and installation requirements, complaint handling, records requirements, servicing requirements and statistical techniques potentially applicable to the production of our medical device product. In addition, we must engage in extensive recordkeeping and reporting and must make available our manufacturing facilities and records for periodic announced or unannounced inspections by governmental agencies, including the FDA, state authorities and comparable agencies in other countries. If we experience an unsuccessful Quality System inspection, our operations could be disrupted and our manufacturing could be interrupted. Failure to take adequate corrective action in response to an adverse Quality System inspection could result in, among other things, a shut-down of our manufacturing operations, significant fines, suspension of marketing clearances and approvals, seizures or recalls of our device, operating restrictions and criminal prosecutions, any of which would cause our business to suffer. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with applicable regulatory requirements, which may result in manufacturing delays for our INVU platform and cause our revenue to decline. We have registered with the FDA as a medical device manufacturer. We anticipate that we and certain of our third-party component suppliers will be subject to FDA and local regulatory inspections.
We or our suppliers may not be able to continue to remain in compliance with QSR. If any manufacturer’s facilities in the United States were found to be in noncompliance or fail to take satisfactory corrective action in response to adverse QSR inspectional findings, the FDA could take legal or regulatory enforcement actions against us and/or our products, including, but not limited to, the cessation of sales or the recall of our products, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be required to bear other costs or take other actions that may have a negative impact on our future sales and our ability to generate profits. Taking corrective action may be expensive, time-consuming and a distraction for management, and if we experience a shutdown or delay at our manufacturing facilities, we may be unable to produce our products, which would harm our business.
Current regulations depend heavily on administrative interpretation. If the FDA does not believe that we are in compliance with applicable FDA regulations, the agency could take legal or regulatory enforcement actions against us and/or our products. We are also subject to periodic inspections and audits by the FDA and other governmental regulatory agencies, as well as certain third-party regulatory groups. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulations occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly and/or time-consuming for us or a third party to implement and that may include the temporary or permanent suspension of a clinical study or commercial sales or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us or third parties with whom we contract could materially harm our business. Future interpretations made by the FDA or other regulatory bodies made during the course of these inspections or audits may vary from current interpretations and may adversely affect our business and prospects. The FDA’s and other comparable non-U.S. regulatory agencies’ statutes, regulations, policies or interpretations may change, and additional government regulation or statutes may be enacted, which could increase post-approval regulatory requirements, or delay, suspend or prevent marketing of any cleared or approved products or necessitate the recall of distributed products. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.
The medical device industry has been under heightened FDA scrutiny as the subject of government investigations and enforcement actions. If our operations and activities are found to be in violation of any FDA laws or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines and other legal and/or agency enforcement actions. Any penalties, damages, fines or curtailment or restructuring of our operations or activities could harm our ability to operate our business and our financial results. The risk of us being found in violation of FDA law or regulations is increased by the fact that many of these laws and regulations are broad and their provisions are open to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend ourselves against that action and its underlying allegations, could cause us to incur significant legal expenses and divert management’s attention from the operation of our business. Where there is a dispute with a federal or state governmental agency that cannot be resolved to the mutual satisfaction of all relevant parties, we may determine that the costs, both real and contingent, are not justified by the commercial returns to us from maintaining the dispute or the product.
Various claims, design features or performance characteristics of our medical device that we may regard as permitted by the FDA without marketing clearance or approval may be challenged by the FDA or state or foreign regulators. The FDA or state or foreign regulatory authorities may find that certain claims, design features or performance characteristics, in order to be made or included in the product, may have to be supported by further studies and marketing clearances or approvals, which could be lengthy, costly and possibly unobtainable.
41
Our INVU platform and wearable wireless sensor band may cause or contribute to adverse medical events or be subject to failures or malfunctions that we are required to report to the FDA, and if we fail to do so, we would be subject to sanctions that could harm our reputation, business, financial condition and results of operations. The discovery of serious safety issues with our wearable wireless sensor band, or a recall of our wearable wireless sensor band either voluntarily or at the direction of the FDA or another governmental authority could have a negative impact on us.
We are subject to the FDA’s medical device reporting regulations and similar foreign regulations, which require us to report to the FDA when we receive or become aware of information that reasonably suggests that one or more of our products may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, it could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of the product. If we fail to comply with our reporting obligations, the FDA could take action, including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device clearance or approval, seizure of our products or delay in clearance or approval of future products.
The FDA and foreign regulatory bodies have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture of a product or in the event that a product poses an unacceptable risk to health. The FDA’s authority to require a recall must be based on a finding that there is reasonable probability that the device could cause serious injury or death. We may also choose to voluntarily recall a product if any material deficiency is found. A government-mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing defects, labeling or design deficiencies, packaging defects or other deficiencies or failures to comply with applicable regulations. Product defects or other errors may occur in the future.
Depending on the corrective action we take to redress a product’s deficiencies or defects, the FDA may require, or we may decide, that we will need to obtain new clearances or approvals for the device before we may market or distribute the corrected device. Seeking such clearances or approvals may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement action, including FDA warning letters, product seizure, injunctions, administrative penalties or civil or criminal fines.
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA. We may initiate voluntary withdrawals or corrections for our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, it could require us to report those actions as recalls and we may be subject to enforcement action. A future recall announcement could harm our reputation with customers, potentially lead to product liability claims against us and negatively affect our sales. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, will distract management from operating our business and may harm our reputation and financial results.
Our products may in the future be subject to product recalls that could harm our reputation, business and financial results.
Medical devices can experience performance problems in the field that require review and possible corrective action. The occurrence of component failures, manufacturing errors, software errors, design defects or labeling inadequacies affecting a medical device could lead to a government-mandated or voluntary recall by the device manufacturer, in particular when such deficiencies may endanger health. The FDA requires that certain classifications of recalls be reported to the FDA within 10 working days after the recall is initiated. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA. We may initiate voluntary recalls in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. Product recalls may divert management attention and financial resources, expose us to product liability or other claims, harm our reputation with customers and adversely impact our business, financial condition and results of operations.
42
Legislative or regulatory reforms may make it more difficult and costly for us to obtain regulatory clearance or approval of any future products and to manufacture, market and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions, which may prevent or delay approval or clearance of our future products under development or impact our ability to modify our currently cleared products on a timely basis. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of planned or future products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be.
FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new statutes, regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of any future products or make it more difficult to obtain clearance or approval for, manufacture, market or distribute our products. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require: additional testing prior to obtaining clearance or approval; changes to manufacturing methods; recall, replacement or discontinuance of our products; or additional record keeping.
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be promulgated that could prevent, limit or delay regulatory clearance or approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. For example, a change in administration may impact our business and industry.
Any change in the laws or regulations that govern the clearance and approval processes relating to our current, planned and future products could make it more difficult and costly to obtain clearance or approval for new products or to produce, market and distribute existing products. Significant delays in receiving clearance or approval or the failure to receive clearance or approval for any new products would have an adverse effect on our ability to expand our business. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing clearance that we may have obtained and we may not achieve or sustain profitability.
From time to time, we engage outside parties to perform services related to certain of our clinical studies and trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to complete our clinical studies and may incur significant additional costs.
From time to time, we engage consultants to help design, monitor and analyze the results of certain of our clinical studies and trials. The consultants we engage interact with clinical investigators to enroll patients in our clinical trials. We depend on these consultants and clinical investigators to conduct clinical studies and trials and monitor and analyze data from these studies and trials under the investigational plan and protocol for the study or trial and in compliance with applicable regulations and standards, such as the FDA’s Good Clinical Practice, or GCP, guidelines and FDA human subject protection regulations. We may face delays in completing our clinical studies if these parties do not perform their obligations in a timely, compliant or competent manner. If these third parties do not successfully carry out their duties or meet expected deadlines, or if the quality, completeness or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical trial protocols or for other reasons, our clinical studies or trials may be extended, delayed or terminated or may otherwise prove to be unsuccessful, and we may have to conduct additional studies, which would significantly increase our costs.
43
Healthcare reform initiatives and other administrative and legislative proposals may harm our business, financial condition, results of operations and cash flows in our key markets.
There have been and continue to be proposals by the federal government, state and local governments, regulators and third-party payers to control or manage the increased costs of healthcare and, more generally, to reform the U.S. healthcare system. Certain of these proposals could limit the prices we are able to charge for our INVU platform or the coverage and reimbursement available for our products and could limit the acceptance and availability of our products. The adoption of proposals to control costs could harm our business, financial condition and results of operations.
There likely will continue to be legislative and regulatory proposals at the federal, state and local levels directed at containing or lowering the cost of healthcare, particularly in light of recent elections. We cannot predict the initiatives that may be adopted in the future or their full impact. The continuing efforts of federal, state and local governments, insurance companies, managed care organizations and other payers of healthcare services to contain or reduce costs of healthcare may harm:
| ● | our ability to set a price that we believe is fair for our product; |
| ● | our ability to generate revenue and achieve or maintain profitability; and |
| ● | the availability of capital. |
Further, heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, has resulted in several U.S. Congressional inquiries and proposed and enacted federal legislation designed to bring transparency to product pricing and reduce the cost of products and services under government healthcare programs. Additionally, individual states in the United States have also increasingly passed legislation and implemented regulations designed to control product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures. Moreover, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what products to purchase and which suppliers will be included in their healthcare programs. Adoption of price controls and other cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures may prevent or limit our ability to generate revenue and attain profitability.
Various new healthcare reform proposals are emerging at the federal and state level. Any new federal, state or local healthcare initiatives that may be adopted could limit the amounts that federal, state or local governments will pay for healthcare products and services, and could harm our business, financial condition and results of operations.
Clinical trials may be delayed, suspended or terminated for many reasons, which will increase our expenses and delay the time it takes to expand our evidence base for our products.
We plan to continue to develop and execute clinical studies for our INVU platform. We may experience delays in ongoing or future clinical studies, and we do not know whether future clinical studies will begin on time, need to be redesigned, enroll an adequate number of patients on time or be completed on schedule, if at all. The commencement and completion of clinical trials for future products or indications may be delayed, suspended or terminated as a result of many factors, including:
| ● | the delay or refusal of regulators or Institutional Review Boards, or IRBs, to authorize us to commence a clinical trial at a prospective trial site; |
| ● | changes in regulatory requirements, policies and guidelines; |
| ● | delays or failure to reach agreement on acceptable terms with prospective clinical research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
44
| ● | delays in patient enrollment and variability in the number and types of patients available for clinical trials; |
| ● | the inability to enroll a sufficient number of patients in trials to observe statistically significant treatment effects in the trial; |
| ● | having clinical sites deviate from the trial protocol or dropping out of a trial; |
| ● | safety or tolerability concerns that could cause us to suspend or terminate a trial if we find that participants are being exposed to unacceptable health risks; |
| ● | regulators or IRBs require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or safety concerns, among others; |
| ● | lower than anticipated retention rates of patients and volunteers in clinical trials; |
| ● | our CROs or clinical trial sites failing to comply with regulatory requirements or meet their contractual obligations to use in a timely manner, or at all, deviating from the protocol or dropping out of a trial; |
| ● | delays relating to adding new clinical trial sites; and |
| ● | exceeding budgeted costs due to difficulty in accurately predicting costs associated with clinical trials. |
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs or the ethics committees of institutions at which such trials are being conducted, by the Data Safety Monitoring Board for such trial or by the FDA or other regulatory authorities. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements, including GCP regulations, or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA resulting in the imposition of a clinical hold, unforeseen safety issues, failure to demonstrate safety and effectiveness, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
In addition, we may encounter delays if the FDA concludes that our financial relationships with investigators result in a perceived or actual conflict of interest that may have affected the interpretation of a study, the integrity of the data generated at the applicable clinical trial site or the utility of the clinical trial itself. Principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time. If these relationships and any related compensation to or ownership interest by the clinical investigator carrying out the study result in perceived or actual conflicts of interest, or if the FDA concludes that the financial relationship may have affected interpretation of the study, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized, which could result in the delay or rejection by the FDA. Any such delay or rejection could prevent us from expanding the evidence base for our INVU platform and may harm future sales growth of our products.
The results of our clinical trials may not support our INVU platform claims or may result in the discovery of adverse side effects.
The results of our future clinical trials may not support our development plans for our INVU platform and the FDA may not agree with our conclusions regarding the results of our trials. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and later trials may not replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our INVU platform is safe and effective for the proposed indicated uses, which could cause us to abandon development of our INVU platform and may delay development of other product candidates. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, our ability to commercialize our INVU platform and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the future product’s profile.
45
Risks Related to Israeli Law and Our Operations in Israel
Conditions in Israel, including the ongoing war between Israel and Hamas, and other conflicts in the region, may adversely affect our business, our results of operations and our ability to raise additional funds.
We are incorporated under the laws of the State of Israel, and many of our employees, including certain management members, operate from our offices that are located in Tel Aviv, Israel. In addition, a number of our officers and directors are residents of Israel. Accordingly, our business and operations are directly affected by economic, political, geopolitical and military conditions in Israel. Since the establishment of the State of Israel in 1948, a number of armed conflicts have occurred between Israel and its neighboring countries and terrorist organizations active in the region. These conflicts have involved missile strikes, hostile infiltrations and terrorism against civilian targets in various parts of Israel, which have negatively affected business conditions in Israel.
In October 2023, Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched extensive rocket attacks on Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel. These attacks resulted in extensive deaths, injuries and kidnapping of civilians and soldiers. Following the attack, Israel’s security cabinet declared war against Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks.
The intensity and duration of Israel’s current war against Hamas is difficult to predict, as are such war’s economic implications on the Company’s business and operations and on Israel’s economy in general. These events may be intertwined with wider macroeconomic indications of a deterioration of Israel’s economic standing, which may have a material adverse effect on the Company and its ability to effectively conduct its operations, for instance, a downgrade in Israel’s credit rating by rating agencies (such as a recent downgrade by Moody’s of its credit rating of Israel from A1 to A2, as well as the downgrade of its outlook rating from “stable” to “negative” and the S&P Global lowered its long-term credit rating from AA- to A+, as well as a downgrade of its short-term credit ratings from A-1+ to A-1, with an outlook on the long-term ratings “negative”).
In connection with the Israeli security cabinet’s declaration of war against Hamas and possible hostilities with other organizations, several hundred thousand Israeli military reservists were drafted to perform immediate military service. Certain of our employees and consultants in Israel, in addition to employees of our service providers located in Israel, have been called, and additional employees may be called, for service in the current or future wars or other armed conflicts with Hamas and others, and such persons may be absent for an extended period of time. As a result, our operations may be disrupted by such absences, which disruption may materially and adversely affect our business and results of operations. Additionally, the absence of employees of our Israeli suppliers and contract manufacturers due to their military service in the current or future wars or other armed conflicts may disrupt their operations, which in turn may materially and adversely affect our ability to deliver or provide products and services to customers.
Following the attack by Hamas on Israel’s southern border, Hezbollah in Lebanon also launched missile, rocket, drone and shooting attacks against Israeli military sites, troops and Israeli towns in northern Israel. In response to these attacked, the Israeli army has carried out a number of targeted strikes on sites belonging to Hezbollah in Lebanon and Syria. Recently, Iran has directly joined the hostilities against Israel by firing hundreds of drones, ballistic missiles and guided missiles to Israel, causing further uncertainty in the region. While currently no damages were registered in Israel from such attack, the situation is developing and could lead to additional wars in the Middle East. It is possible that other terrorist organizations, including Palestinian military organizations in the West Bank, as well as other hostile countries, will join the hostilities. Such hostilities may include terror and missile attacks. In the event that our facilities are damaged as a result of hostile actions, or hostilities otherwise disrupt our ongoing operations, our ability to deliver or provide products and services in a timely manner to meet our contractual obligations towards customers and vendors could be materially and adversely affected. Our commercial insurance does not cover losses that may occur as a result of events associated with war and terrorism. Although the Israeli government currently covers the reinstatement value of certain direct damages that are caused by terrorist attacks or acts of war, we cannot assure you that such government coverage will be maintained or that it will sufficiently cover our potential damages. Any losses or damages incurred by us could have a material adverse effect on our business.
46
In addition, some countries around the world restrict doing business with Israel and Israeli companies, and additional countries may impose restrictions on doing business with Israel and Israeli companies if hostilities in Israel or political instability in the region continue or increase. These restrictions may limit materially our ability to sell our products and provide our services to companies and customers in these countries. In addition, there have been increased efforts by activists to cause companies and consumers to boycott Israeli goods and services. Such efforts, particularly if they become more widespread, may materially and adversely impact our ability to sell and provide our products and services outside of Israel.
Furthermore, following Hamas’ attack on Israel and Israel’s security cabinet declaration of war against Hamas, the Houthi movement, which controls parts of Yemen, launched a number of attacks on marine vessels traversing the Red Sea, which marine vessels were thought to either be in route towards Israel or to be partly owned by Israeli businessmen. The Red Sea is a vital maritime route for international trade traveling to or from Israel. As a result of such disruptions, we may experience in the future delays in supplier deliveries, extended lead times, and increased cost of freight, increased insurance costs, purchased materials and manufacturing labor costs. The risk of ongoing supply disruptions may further result in delayed deliveries of our products.
Prior to the Hamas attack in October 2023, the Israeli government pursued extensive changes to Israel’s judicial system, which sparked extensive political debate and unrest. In response to such initiative, many individuals, organizations and institutions, both within and outside of Israel, have voiced concerns that the proposed changes may negatively impact the business environment in Israel including due to reluctance of foreign investors to invest or transact business in Israel, as well as to increased currency fluctuations, downgrades in credit rating, increased interest rates, increased volatility in security markets and other changes in macroeconomic conditions. The risk of such negative developments has increased in light of the recent Hamas attacks and the war against Hamas declared by Israel, regardless of the proposed changes to the judicial system and the related debate. To the extent that any of these negative developments do occur, they may have an adverse effect on our business, our results of operations and our ability to raise additional funds, if deemed necessary by our management and board of directors
It may be difficult to enforce a U.S. judgment against us, and our officers and directors named in this Annual Report, in Israel or the United States, or to assert U.S. securities law claims in Israel or serve process on our officers and directors.
Not all of our directors or officers are residents of the United States and most of their and our assets are located outside the United States. Service of process upon us or our non-U.S. resident directors and officers and enforcement of judgments obtained in the United States against us or our non-U.S. directors and executive officers may be difficult to obtain within the United States. We have been informed by our legal counsel in Israel that it may be difficult to assert claims under U.S. securities law in original actions instituted in Israel or obtain a judgment based on the civil liability provisions of U.S. federal securities law. Israeli courts may refuse to hear a claim based on a violation of U.S. securities law against us or our non-U.S. officers and directors because Israel may not be the most appropriate forum to bring such a claim. In addition, even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by Israeli law. There is little binding case law in Israel addressing the matters described above. Under certain circumstances, Israeli courts might not enforce judgments rendered outside Israel, which may make it difficult to collect on judgments rendered against us or our non-U.S. officers and directors.
47
We have received Israeli government grants for certain research and development activities. The terms of those grants require us to satisfy specified conditions as defined in Israel’s Encouragement of Research, Development and Technological Innovation in Industry Law, 5744-1984, or Innovation Law. Such grants will restrict the transfer or license of know-how, and may restrict the transfer of manufacturing or manufacturing rights of certain products, technologies or know-how outside of Israel, without the prior approval of the IIA.
Our research and development efforts were financed, in part, through grants from the Israel Innovation Authority, or IIA. From our inception, we conducted projects with the IIA’s support and received from the IIA grants totaling, as of December 31, 2023, approximately $1.164 million. The grants received from the IIA before June 30, 2017, in the amount of approximately $1,030,00 bear an annual interest rate that applied at the time of the approval of the applicable file and such interest will apply to all the funding received under that approval. The grant received from the IIA after June 30, 2017, in the amount of approximately $81,417 bear an annual interest rate based on the 12-month London Interbank Offered Rate, or LIBOR until December 31, 2023, and as of January 1, 2024, bear an annual interest rate based on the 12-month Secured Overnight Financing Rate, or the SOFR, or in an alternative publication by the Bank of Israel, with the addition of 0.71%.
Pursuant to the Innovation Law and the regulations thereunder, we will be required to return the grants by a payment of royalties at a rate of 3% to 5% on sales proceeds from our products that we developed under IIA programs up to the total amount of grants received, linked to the U.S. dollar and bearing interest rate at SOFR, applicable to U.S. dollar deposits, as published on the first business day of each calendar year.
Even after full repayment of the grants received, the Innovation Law requires, inter alia, that the products developed as part of the programs under which the grants were given be manufactured in Israel and restricts the ability to transfer know-how funded by the IIA outside of Israel. Transfer of IIA-funded know-how outside of Israel requires prior approval and is subject to payment of a redemption fee to the IIA calculated according to a formula provided under the Innovation Law. A transfer for the purpose of the Innovation Law is generally interpreted very broadly and includes, inter alia, any actual sale of the IIA-funded know-how, any license to manufacture or develop the IIA-funded know-how or the products resulting from such IIA-funded know-how or any other transaction, which, in essence, constitutes a transfer of IIA-funded know-how. We cannot be certain that any approval of the IIA will be obtained on terms that are acceptable to us, or at all. We may not receive the required approvals should we wish to transfer IIA-funded know-how and/or development outside of Israel in the future.
Subject to prior approval of the IIA, we may transfer the IIA-funded know-how to another Israeli company. If the IIA-funded know-how is transferred to another Israeli entity, the transfer would still require IIA approval but will not be subject to the payment of the redemption fee. In such case, the Israeli acquiring company would have to assume all of the applicable restrictions and obligations towards the IIA (including the restrictions on the transfer of know-how and manufacturing capacity, to the extent applicable, outside of Israel) as a condition to IIA approval.
We may become subject to claims for remuneration or royalties for assigned service invention rights by our employees, which could result in litigation and adversely affect our business.
A significant portion of our intellectual property has been developed by our employees in the course of their employment for us. Under the Israeli Patent Law, 5727-1967, or the Patent Law, inventions conceived by an employee in the course and as a result of or arising from his or her employment with a company are regarded as “service inventions,” which belong to the employer, absent a specific agreement between the employee and employer giving the employee service invention rights. The Patent Law also provides that if there is no such agreement between an employer and an employee, the Israeli Compensation and Royalties Committee (the “Committee”), a body constituted under the Patent Law, shall determine whether the employee is entitled to remuneration for his or her inventions. Case law clarifies that the right to receive consideration for “service inventions” can be waived by the employee. The Committee will examine, on a case-by-case basis, the general contractual framework between the parties, using interpretation rules of the general Israeli contract laws. Further, the Committee has not yet determined one specific formula for calculating this remuneration, but rather uses the criteria specified in the Patent Law. Although we generally enter into assignment-of-invention agreements with our employees pursuant to which such individuals waive their right to remuneration for service inventions, we may face claims demanding remuneration in consideration for assigned inventions. As a consequence of such claims, we could be required to pay additional remuneration or royalties to our current and/or former employees, or be forced to litigate such claims, which could negatively affect our business.
48
Exchange rate fluctuations between the U.S. dollar, Euro and the New Israeli Shekel currencies may negatively affect our earnings.
Our functional currency is the U.S. dollar. We incur expenses in U.S. dollars, Euros and NIS. As a result, we are exposed to the risks that the Euro and the NIS may depreciate relative to the U.S. dollar, or, if either the Euro and the NIS devalue relative to the U.S. dollar, that the inflation rate in the EU and in Israel may exceed such rate of devaluation of the Euro and the NIS, or that the timing of such devaluation may lag behind inflation in the EU and in Israel. In any such event, the Euro-denominated cost of our operations in the EU and our NIS denominated costs of our operations in Israel would increase and our U.S. dollar-denominated results of operations would be adversely affected. The average exchange rate for the year ended December 31, 2023 was $1.00 = Euro 0.924 and $1.00 = NIS 3.687. We cannot predict any future trends in the rate of inflation in the EU and in the United States or the rate of devaluation, if any, of either the Euro or the U.S. dollar against the NIS.
If we are not able to successfully hedge against the risks associated with currency fluctuations, our financial condition and results of operations could be adversely affected. While we may decide to enter into hedging transactions in the future, the availability and effectiveness of these hedging transactions may be limited and we may not be able to successfully hedge our exposure, which could adversely affect our financial condition and results of operations.
The termination or reduction of tax and other incentives that the Israeli government provides to Israeli companies may increase our costs and taxes.
The Israeli government currently provides tax and capital investment incentives to Israeli companies, as well as grant and loan programs relating to research and development and marketing and export activities. In recent years, the Israeli government has reduced the benefits available under these programs and the Israeli governmental authorities may in the future further reduce or eliminate the benefits of these programs. We may take advantage of these benefits and programs in the future; however, there can be no assurance that such benefits and programs will be available to us. If we qualify for such benefits and programs and fail to meet the conditions thereof, the benefits could be canceled and we could be required to refund any benefits we might already have enjoyed, including interest and linkage difference, and become subject to penalties. Additionally, if we qualify for such benefits and programs and they are subsequently terminated or reduced, it could have an adverse effect on our financial condition and results of operations.
49
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain patent or other intellectual property protection for any product we develop or for our technology, or if the scope of the patent and other intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize products and technology similar or identical to ours, and our ability to successfully commercialize any product we may develop, and our technology, may be harmed.
As with other medical device companies, our success depends in large part on our ability to obtain, maintain and solidify a proprietary position for our product, which will depend upon our success in obtaining effective patent protection in the United States and other countries that cover, and other intellectual property with respect to, such product, its manufacturing processes and its intended methods of use and enforcing those patent claims once granted, as well as our other intellectual property. In some cases, we may not be able to obtain issued claims covering our technologies which are sufficient to prevent third parties, such as our competitors, from utilizing our technology. Any failure to obtain or maintain patent and other intellectual property protection with respect to our products or other aspects of our business could harm our business, financial condition and results of operations.
Changes in either the patent laws or their interpretation in the United States and other countries may diminish our ability to protect our inventions, obtain, maintain and enforce our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our patents. Additionally, we cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors or other third parties.
The patent prosecution process is expensive, time-consuming and complex, and we may not be able to file, prosecute, maintain, enforce or license all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output in time to obtain patent protection. Although we enter into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, such as our employees, corporate collaborators, outside scientific collaborators, suppliers, consultants, advisors and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection.
The strength of patent rights generally, and particularly the patent position of medical device companies, involves complex legal and scientific questions and can be uncertain, and has been the subject of much litigation in recent years. This uncertainty includes changes to the patent laws through either legislative action to change statutory patent law or court action that may reinterpret existing law or rules in ways affecting the scope or validity of issued patents. Our current or future patent applications may fail to result in issued patents in the United States or foreign countries with claims that cover our product. Even if patents do successfully issue from our patent applications, third parties may challenge the validity, enforceability or scope of such patents, which may result in such patents being narrowed, invalidated or held unenforceable. Any successful challenge to our patents could deprive us of exclusive rights necessary for the successful commercialization of our product. Furthermore, even if they are unchallenged, our patents may not adequately protect our product, provide exclusivity for our products or prevent others from designing around our claims. If the scope of any patent protection we obtain is not sufficiently broad, or if we lose any of our patent protection, our ability to prevent our competitors from commercializing similar or identical technology and products would be adversely affected. If the breadth or strength of protection provided by the patents we hold or pursue with respect to our products is challenged, it could dissuade companies from collaborating with us to develop, or threaten our ability to commercialize, our product.
Patent terms may be inadequate to protect our competitive position on our future products for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our future products are obtained, once the patent life has expired, we may be open to competition from competitive products.
Given the amount of time required for the development, testing and regulatory review of new products, patents protecting our future products might expire before or shortly after we or our future partners commercialize those products. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours for a sufficient amount of time, and, as a result, we may not be able to obtain adequate protection from our patent portfolio against competition, in spite of the time and effort invested in the commercialization of our future products.
50
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time-consuming and unsuccessful, and which could additionally result in the diversion of management’s time and efforts, require us to pay damages or prevent us from marketing our existing or future products.
Competitors may infringe our patents, or the patents of any future licensing partners, or we may be required to defend against claims of infringement. In addition, our patents or the patents of any such licensing partners also may become involved in inventorship, priority or validity disputes. Patent litigation is prevalent in the medical device and diagnostic sectors. Our commercial success depends in part upon our ability and that of our contract manufacturers and suppliers to manufacture, market, sell our planned product, and use our proprietary technologies without infringing, misappropriating or otherwise violating the proprietary rights or intellectual property of third parties. To prosecute, counter or defend against patent infringement claims can be expensive and time-consuming. In an infringement proceeding, a court may decide that our patent is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover such technology. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during litigation.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our management and other personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial negative impact on the price of Holdco Ordinary Shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could harm our ability to compete in the marketplace. Any of the foregoing could harm our business, financial condition and results of operations.
We may in the future become party to, or be threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology. Additional third parties may assert infringement claims against us based on existing or future intellectual property rights, regardless of merit. If we are found to infringe a third-party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our products and technology. We may also elect to enter into such a license in order to settle pending or threatened litigation. However, we may not be able to obtain any required license on commercially reasonable terms, or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us, and could require us to pay significant royalties and other fees. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, which may be significant. If we are found to have willfully infringed a third-party patent, we could be required to pay treble damages and attorneys’ fees. A finding of infringement could prevent us from commercializing our planned products in commercially important territories, or force us to cease some of our business operations, which could harm our business. Although we try to ensure that our employees, advisors and consultants do not use the proprietary information or know-how of others in their work for us, including requiring that our employees, advisors and consultants do not breach any contractual obligations with other third parties related to proprietary information or know-how in connection with their work for us, we may be subject to claims that we, or these employees, have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. These and other claims that we have misappropriated the confidential information or trade secrets of third parties can have a similar negative impact on our business to the infringement claims discussed above.
Even if we are successful in prosecuting or defending against intellectual property claims, litigation or other legal proceedings relating to such claims may cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial negative impact on the price of Holdco Ordinary Shares. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can. Uncertainties resulting from the initiation and continuation of litigation or other intellectual property related proceedings could harm our business, financial condition and results of operations.
51
We have foreign intellectual property rights and may not be able to protect our intellectual property and proprietary rights throughout the world, which could harm our business, financial condition and results of operations.
Currently, our technology is protected by an extensive global patent portfolio consisting of 43 issued foreign utility patents, one allowed foreign utility patent application, 13 pending foreign utility patent applications and one PCT patent application. Filing, prosecuting and defending patents on our products in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, from selling or importing products made using our inventions in and into the United States or other jurisdictions, or from using our trademarks or trademarks confusingly similar to our trademarks for similar goods and services in any jurisdiction. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our product, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our intellectual property and proprietary rights generally. Proceedings to enforce our intellectual property and proprietary rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property and proprietary rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition and results of operations may be harmed.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be harmed.
Our registered and unregistered trademarks or trade names may be challenged, infringed, circumvented, declared generic or determined to be violating or infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential partners and customers in our markets of interest. At times, competitors or other third parties may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement or dilution claims brought by owners of other trademarks. We have not conducted any registrability studies for possible future trademarks or current trademarks in future jurisdictions to assess whether such marks would be successfully registered. In addition, we may license our trademarks and trade names to third parties, such as distributors. Although these license agreements may provide guidelines for how our trademarks and trade names may be used, a breach of these agreements or misuse by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, trade secrets, domain names or other intellectual property may be ineffective, and we may be required, and have been required, to limit the use of our trademarks or trade names to certain classes of goods and services in certain jurisdictions. This could result in substantial costs and diversion of resources and could harm our business, financial condition and results of operations.
52
Our use of “open source” software could subject our proprietary software to general release, adversely affect our ability to sell our products and subject us to possible litigation.
A portion of the products or technologies licensed, developed and/or distributed by us incorporate so-called “open source” software and we may incorporate open source software into other products and technologies in the future. Such open source software generally is licensed by its authors or other third parties under open source licenses. Some open source licenses may contain certain unfavorable conditions, such as requirements that we disclose source code for modifications or derivative works that we make to the open source software and that we license such modifications or derivative works to third parties at no cost or under the terms of the particular open source license. In some circumstances, distribution of our proprietary software in connection with open source software could require that we disclose and license some or all of our proprietary code in that software, as well as distribute our software that uses particular open source software at no cost to the user. We monitor our use of open source software in an effort to avoid uses in a manner that would require us to disclose or grant licenses under our proprietary source code; however, there can be no assurance that such efforts will be successful. In addition, third-party products that we utilize may also include certain open source software code that if used in combination with our own software may jeopardize our intellectual property rights or limit our ability to sell through certain sales channels (or otherwise subject our proprietary source code to the above-described risks). Open source license terms are often ambiguous and such use could inadvertently occur. There is little legal precedent governing the interpretation of many of the terms of these licenses, and the potential impact of these terms on our business may result in unanticipated obligations regarding our technologies. Companies that incorporate open source software into their products have, in the past, faced claims seeking enforcement of open source license provisions and claims asserting ownership of open source software incorporated into their product. If an author or other third party that distributes such open source software were to allege that we had not complied with the conditions of an open source license, we could incur significant legal costs defending ourselves against such allegations. In the event such claims were successful, we could be subject to significant damages or be enjoined from the distribution of the infringing product. In addition, if we combine our proprietary software with open source software in certain ways, under some open source licenses, we could be required to release the source code of our proprietary software, which could substantially help our competitors develop products that are similar to or better than ours and otherwise adversely affect our business. These risks could be difficult to eliminate or manage, and, if not addressed, could harm our business, financial condition and results of operations.
53
Risks Related to the Ownership of the Holdco Ordinary Shares
There can be no assurance that we will be able to comply with the continued listing standards of Nasdaq.
The Holdco Ordinary Shares and Holdco Warrants on Nasdaq are currently traded on Nasdaq. If Holdco subsequently does not continue to satisfy any additional listing standards, Holdco and its shareholders could face significant material adverse consequences including:
| ● | a limited availability of market quotations for its securities; |
| ● | reduced liquidity for its securities; |
| ● | a determination that the Holdco Ordinary Shares are a “penny stock” which will require brokers trading in the Holdco Ordinary Shares to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for its securities; |
| ● | a limited amount of news and analyst coverage; and |
| ● | a decreased ability to issue additional securities or obtain additional financing in the future. |
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because the Holdco Ordinary Shares are listed on Nasdaq, they are covered securities. Although the states are preempted from regulating the sale of Holdco’s securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. While Holdco is not aware of a state, other than the State of Idaho, having used these powers to prohibit or restrict the sale of securities issued by blank check companies, certain state securities regulators view blank check companies unfavorably and might use these powers, or threaten to use these powers, to hinder the sale of securities of blank check companies in their states.
An active market for Holdco Ordinary Shares may not develop, which would adversely affect the liquidity and price of Holdco Ordinary Shares.
An active trading market for Holdco Ordinary Shares may never develop or, if developed, it may not be sustained. You may be unable to sell your Holdco Ordinary Shares unless an active trading market can be established and sustained.
Holdco Warrants will become exercisable for Holdco Ordinary Shares, which would increase the number of shares eligible for future resale in the public market and result in dilution to Holdco’s shareholders.
Outstanding Holdco Warrants to purchase an aggregate of 13,223,440 Holdco Ordinary Shares, which were formerly LAMF Warrants, will become exercisable in accordance with the terms of the Warrant Assignment, Assumption and Amendment Agreement governing those securities 30 days after the Closing. The exercise price of such warrants will be $11.50 per share. To the extent such warrants are exercised, additional Holdco Ordinary Shares will be issued, which will result in dilution to the holders of Holdco Ordinary Shares and increase the number of Holdco Ordinary Shares eligible for resale in the public market. Sales of substantial numbers of such Holdco Ordinary Shares in the public market or the fact that such warrants may be exercised could adversely affect the market price of Holdco Ordinary Shares. However, there is no guarantee that such warrants will ever be in the money prior to their expiration, and as such, the warrants may expire worthless.
54
If Holdco does not meet the expectations of equity research analysts, if they do not publish research or reports about Holdco’s business or if they issue unfavorable commentary or downgrade Holdco Ordinary Shares, the price of Holdco Ordinary Shares could decline.
The price of Holdco Ordinary Shares and trading volume may be heavily influenced by the way analysts and investors interpret our financial information and other disclosures. If securities or industry analysts do not publish research or reports about our business, delay publishing reports about our business, or publish negative reports about our business, regardless of accuracy, the market price and trading volume of Holdco Ordinary Shares could decline.
If an active trading market for Holdco Ordinary Shares develops, the trading market will be influenced to some extent by the research and reports that industry or financial analysts publish about us and our business. We have little to no influence over these analysts. As a newly public company, we may be slow to attract research coverage and the analysts who publish information about Holdco Ordinary Shares will have had relatively little experience with us or our industry, which could affect their ability to accurately forecast our results and could make it more likely that we fail to meet their estimates. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us provide inaccurate or unfavorable research or issue an adverse opinion regarding Holdco Ordinary Shares, the price of Holdco Ordinary Shares could decline. The share prices of many companies in the technology industry have declined significantly after those companies have failed to meet, or significantly exceed, the financial guidance they have publicly announced or the expectations of analysts and investors. If our financial results fail to meet, or significantly exceed, our announced guidance or the expectations of analysts or investors, analysts could downgrade Holdco Ordinary Shares or publish unfavorable research about us. Furthermore, if one or more of these analysts cease coverage of us or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our share price or trading volume to decline.
Even if Holdco Ordinary Shares are actively covered by analysts, we do not have any control over the analysts or the measures that analysts or investors may rely upon to forecast our future results. Over-reliance by analysts or investors on any particular metric to forecast our future results may lead to forecasts that differ significantly from our own.
The price of Holdco Ordinary Shares may be volatile.
The price of the Holdco Ordinary Shares may fluctuate due to a variety of factors, including:
| ● | changes in the industries in which Holdco and its customers operate; |
| ● | developments involving Holdco’s competitors; |
| ● | changes in laws and regulations affecting its business; |
| ● | variations in its operating performance and the performance of its competitors in general; |
| ● | actual or anticipated fluctuations in Holdco’s quarterly or annual operating results; |
| ● | publication of research reports by securities analysts about Holdco or its competitors or its industry; |
| ● | the public’s reaction to Holdco’s press releases, its other public announcements and its filings with the SEC; |
| ● | actions by shareholders; |
| ● | additions and departures of key personnel; |
| ● | commencement of, or involvement in, litigation involving the combined company; |
55
| ● | changes in its capital structure, such as future issuances of securities or the incurrence of additional debt; |
| ● | the volume of Holdco Ordinary Shares available for public sale; and |
| ● | general economic and political conditions, such as the Russia – Ukraine conflict, recessions, interest rates, local and national elections, fuel prices, international currency fluctuations, corruption, political instability and acts of war or terrorism, and a resurgence of the COVID-19 outbreak. |
These market and industry factors may materially reduce the market price of Holdco Ordinary Shares regardless of the operating performance of Holdco.
Certain recent public offerings of companies with public floats comparable to Holdco public float have experienced extreme volatility that was seemingly unrelated to the underlying performance of the respective company. Holdco may experience similar volatility, which may make it difficult for prospective investors to assess the value of the Holdco Ordinary Shares.
Holdco Ordinary Shares may be subject to extreme volatility that is seemingly unrelated to the underlying performance of our business. Recently, companies with comparable public floats and initial public offering sizes have experienced instances of extreme stock price run-ups followed by rapid price declines, and such stock price volatility was seemingly unrelated to the respective company’s underlying performance. Although the specific cause of such volatility is unclear, our public float may amplify the impact of the actions taken by a few shareholders on the price of Holdco Ordinary Shares, which may cause our share price to deviate, potentially significantly, from a price that better reflects the underlying performance of our business. Should Holdco Ordinary Shares experience run-ups and declines that are seemingly unrelated to Holdco’s actual or expected operating performance and financial condition or prospects, prospective investors may have difficulty assessing the rapidly changing value of Holdco Ordinary Shares. In addition, investors in Holdco Ordinary Shares may experience losses, which may be material, if the price of Holdco Ordinary Shares declines after the Closing or if such investors purchase Ordinary Shares prior to any price decline.
It is not expected that Holdco will pay dividends in the foreseeable future.
Holdco expects to retain most, if not all, of its available funds and any future earnings after the Business Combination to fund the development and growth of its business. As a result, it is not expected that Holdco will pay any cash dividends in the foreseeable future.
The Holdco Board will have complete discretion as to whether to distribute dividends. Even if the Holdco Board decides to declare and pay dividends, the timing, amount and form of future dividends, if any, will depend on the future results of operations and cash flow, capital requirements and surplus, the amount of distributions, if any, received by Holdco from subsidiaries, Holdco’s financial condition, contractual restrictions and other factors deemed relevant by the Holdco Board. There is no guarantee that the Holdco Ordinary Shares will appreciate in value or that the trading price of the Holdco Ordinary Shares will not decline.
We may issue additional Holdco Ordinary Shares or Holdco Preferred Shares under the Amended Articles, which would dilute the interest of Holdco’s shareholders.
The Amended Articles authorize the issuance of 500,000,000 Holdco Ordinary Shares and 10,000,000 Holdco Preferred Shares. Holdco may issue a substantial number of additional Holdco Ordinary Shares or Holdco Preferred Shares under the Amended Articles. The issuance of additional Holdco shares:
| ● | may significantly dilute the equity interest of investors, who will not have preemption rights in respect of such an issuance; |
| ● | may subordinate the rights of holders of Holdco Ordinary Shares if one or more classes of preferred shares are created, and such preferred shares are issued, with rights senior to those afforded to Holdco Ordinary Shares; |
| ● | could cause a change in control if a substantial number of Holdco Ordinary Shares are issued, which may affect, among other things, our ability to use our net operating loss carry forwards, if any, and could result in the resignation or removal of our present officers and directors; and |
| ● | may adversely affect prevailing market prices for Holdco Ordinary Shares and/or Holdco Warrants. |
56
Holdco may not be able to timely and effectively implement controls and procedures required by Section 404(a) of the Sarbanes-Oxley Act.
Holdco is required to provide management’s attestation on internal controls in connection with Holdco’s second annual report on Form 20-F following consummation of the Business Combination. The standards required for a public company under Section 404(a) of the Sarbanes-Oxley Act are significantly more stringent than those required of Nuvo as a privately-held company. Management may not be able to effectively and timely implement controls and procedures that adequately respond to the increased regulatory compliance and reporting requirements. If Holdco is not able to implement the additional requirements of Section 404(a) in a timely manner or with adequate compliance, it may not be able to assess whether its internal controls over financial reporting are effective, which may subject it to adverse regulatory consequences and could harm investor confidence and the market price of Holdco Ordinary Shares.
As a foreign private issuer and a company treated as an emerging growth company for certain purposes, Holdco has different disclosure and other requirements than U.S. domestic registrants and non-emerging growth companies.
As a foreign private issuer and a company treated as an emerging growth company for certain purposes, Holdco is subject to different disclosure and other requirements than domestic U.S. registrants and non-emerging growth companies. For example, as a foreign private issuer, in the United States, Holdco is not subject to the same disclosure requirements as a domestic U.S. registrant under the Exchange Act, including the requirements to prepare and issue quarterly reports on Form 10-Q or to file current reports on Form 8-K upon the occurrence of specified significant events, the proxy rules applicable to domestic U.S. registrants under Section 14 of the Exchange Act or the insider reporting and short-swing profit rules applicable to domestic U.S. registrants under Section 16 of the Exchange Act. In addition, Holdco is relying on exemptions from certain U.S. rules which permit Holdco to follow Israeli legal requirements rather than certain of the requirements that are applicable to U.S. domestic registrants. However, the laws and regulations of the State of Israel do not contain any provisions applicable to Holdco that are comparable to the U.S. proxy rules, the U.S. rules relating to the filing of reports on Form 10-Q or 8-K or the U.S. rules relating to liability for insiders who profit from trades made in a short period of time.
Furthermore, foreign private issuers are required to file their annual report on Form 20-F within 120 days after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from Regulation Fair Disclosure (“Regulation FD”), aimed at preventing issuers from making selective disclosures of material information, although Holdco is subject to Israeli laws and regulations having substantially the same effect as Regulation FD. As a result of the above, even though Holdco is required to file reports on Form 6-K disclosing the limited information which Holdco has made or is required to make public pursuant to Israeli law, or is required to distribute to shareholders generally, and that is material to Holdco, you may not receive information of the same type or amount that is required to be disclosed to shareholders of a U.S. company.
The JOBS Act contains provisions that, among other things, relax certain reporting requirements for emerging growth companies. Under this Act, as a company treated as an emerging growth company for certain purposes, Holdco is not subject to the same disclosure and financial reporting requirements as non-emerging growth companies. For example, Holdco is permitted to, and intends to take advantage of, certain exemptions that allow it to comply with reduced disclosure obligations in this Annual Report that are applicable to other public companies that are not emerging growth companies. As a result, its shareholders may not have access to certain information that they deem important. Accordingly, the information about Holdco available to you will not be the same as, and may be more limited than, the information available to shareholders of a non-emerging growth company.
Holdco is not required to file periodic reports and financial statements with the SEC as frequently or within the same time frames as U.S. companies with securities registered under the Exchange Act.
Holdco cannot predict if investors will find Holdco Ordinary Shares less attractive because Holdco relies on these exemptions. If some investors find Holdco Ordinary Shares less attractive as a result, there may be a less active trading market for Holdco Ordinary Shares and the share price may be more volatile.
57
Holdco may lose its foreign private issuer status, which would then require Holdco to comply with the Exchange Act’s domestic reporting regime and cause Holdco to incur significant legal, accounting and other expenses.
The determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter. In order to maintain its current status as a foreign private issuer, either (a) more than 50% Holdco’s Ordinary Shares must be either directly or indirectly owned of record by non-residents of the United States or (b)(1) a majority of Holdco’s executive officers or directors must not be U.S. citizens or residents; (2) more than 50% of Holdco’s assets must be located outside of the United States; and (3) Holdco’s business must be administered principally outside the United States. If Holdco loses this status, Holdco would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. Holdco may also be required to make changes in its corporate governance practices in accordance with various SEC and Nasdaq rules. The regulatory and compliance costs to Holdco under U.S. securities law if it is required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the costs Holdco will incur as a foreign private issuer.
Estimates of market opportunity and forecasts of market growth may prove to be inaccurate, and even if the market in which Holdco competes achieves the forecasted growth, Holdco’s business could fail to grow at similar rates, if at all.
Market opportunity estimates and growth forecasts are subject to significant uncertainty and are based on assumptions and estimates that may not prove to be accurate. The variables that go into the calculation of Holdco’s market opportunity are subject to change over time, and there is no guarantee that any particular number or percentage of companies covered by Holdco’s market opportunity estimates will purchase Holdco’s products at all or generate any particular level of revenue for Holdco. Any expansion in Holdco’s market depends on a number of factors, including the cost, performance, and perceived value associated with Holdco’s platform and products and those of its competitors. Even if the market in which Holdco competes meets the size estimates and growth forecasted, Holdco’s business could fail to grow at similar rates, if at all. Holdco’s growth is subject to many factors, including its success in implementing its growth strategies, which are subject to many risks and uncertainties. Accordingly, Holdco’s forecasts of market growth should not be taken as indicative of its future growth.
The Amended Articles provide that, unless Holdco consents to an alternative forum, the federal district courts of the United States shall be the exclusive forum for resolution of any complaint asserting a cause of action arising under the Securities Act, and the competent courts of Tel Aviv, Israel, shall be the exclusive forum for resolution of substantially all disputes between Holdco and its shareholders under the Amended Articles, the Companies Law and the Israeli Securities Law, as well as any derivative action brought on behalf of Holdco and any claim of breach of fiduciary duty owed by a director, officer or other employee of Holdco, which could limit Holdco’s shareholders’ ability to choose the judicial forum for disputes with Holdco, its directors, shareholders, or other employees and could result in increased costs to such shareholders who bring a claim.
Section 22 of the Securities Act creates concurrent jurisdiction for U.S. federal and state courts over all such Securities Act actions. Accordingly, both U.S. state and federal courts have jurisdiction to entertain such claims. To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, the Amended Articles provide that, unless Holdco consents in writing to the selection of an alternative forum, the federal district courts of the United States shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. This exclusive forum provision will not apply to suits brought to enforce any liability or duty created by the Exchange Act and Holdco’s shareholders cannot and will not be deemed to have waived Holdco’s compliance with the U.S. federal securities law and the rules and regulations thereunder as a result of the exclusive forum provision.
The Amended Articles further provide that, unless Holdco consents in writing to the selection of an alternative forum, the competent courts of Tel Aviv, Israel, shall be the exclusive forum for the resolution of (i) any derivative action or proceeding brought on behalf of Holdco, (ii) any action asserting a claim of breach of fiduciary duty owed by any of Holdco’s directors, officers or other employees to Holdco or its shareholders, or (iii) any action asserting a claim arising pursuant to any provision of the Amended Articles, the Companies Law or the Israeli Securities Law, 1968 (the “Israeli Securities Law”). Such exclusive forum provision is intended to apply to claims arising under Israeli law and shall not apply to claims for which the federal courts would have exclusive jurisdiction, whether by law or pursuant to the Amended Articles, as described above.
58
Any person or entity purchasing or otherwise acquiring any interest in any of Holdco’s securities shall be deemed to have notice of and consented to the foregoing provisions of the Amended Articles. However, the enforceability of similar forum provisions (including exclusive federal forum provisions for actions, suits or proceedings asserting a cause of action arising under the Securities Act) in other companies’ organizational documents has been challenged in legal proceedings, and there is uncertainty as to whether courts would enforce the exclusive forum provisions in the Amended Articles. If a court were to find the exclusive forum provisions contained in the Amended Articles to be inapplicable or unenforceable in an action, Holdco may incur additional costs associated with resolving such action in other jurisdictions, which could materially adversely affect Holdco’s business, financial condition and results of operations.
This choice of forum provision may increase costs, such as those related to legal fees and transportation for counsel and the plaintiff, to shareholders by requiring litigating in the courts provided by the exclusive forum provisions in lieu of a more convenient and cost effective jurisdiction for the plaintiff, which may discourage lawsuits against Holdco and its directors, officers, and employees. The exclusive forum provisions could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees. Although we believe these exclusive forum provisions benefit us by providing increased consistency in the application of U.S. federal securities law or the Companies Law or Israeli Securities Law, as applicable, in the types of lawsuits to which they apply, such exclusive forum provisions may limit the shareholders’ abilities to bring a claim in the judicial forum of their choosing for disputes with Holdco or any of its directors, shareholders, officers, or other employees, which may discourage lawsuits with respect to such claims against Holdco and its current and former directors, shareholders, officers or other employees and result in increased costs to such shareholders who bring a claim.
Provisions of Israeli law and the Amended Articles may delay, prevent or make undesirable an acquisition of all or a significant portion of our shares or assets.
Provisions of Israeli law and the Amended Articles which became effective upon the consummation of the Business Combination could have the effect of delaying or preventing a change in control and may make it more difficult for a third party to acquire Holdco or its shareholders to elect different individuals to the Holdco Board, even if doing so would be considered to be beneficial by some of the Holdco Shareholders, and may limit the price that investors may be willing to pay in the future for Holdco Ordinary Shares. The amendments consist of, among other things:
| ● | the Companies Law regulates mergers and requires that a tender offer be effected when one or more persons or entities propose to purchase shares that would result in it or them owning more than a specified percentage of shares in a company; |
| ● | the Companies Law requires special approvals for certain transactions involving directors, officers or significant shareholders and regulates other matters that may be relevant to these types of transactions; |
| ● | the Companies Law does not provide for shareholder action by written consent for public companies, thereby requiring all shareholder actions to be taken at a general meeting of shareholders; |
| ● | the Amended Articles do not permit the Company to undertake a Deemed Liquidation Event, other than a Qualified Deemed Liquidation (each as defined in the Amended Articles) without the consent of the holders of at least a majority of the voting power represented by the then issued and outstanding Holdco Preferred Shares (the “Preferred Majority”); |
| ● | the Amended Articles establish a liquidation and dividend preference for the holders of Holdco Preferred Shares, such upon such an event, such holders are entitled to receive, from the assets available for distribution to the shareholders, prior and in preference to any distribution in respect of Holdco Ordinary Shares, an amount per Holdco Preferred Share equal to the greater of (i) three times the Original Issue Price of such share (as adjusted for certain recapitalization events) or (ii) the amount that would be received if such share had converted into a Holdco Ordinary Share immediately prior to the distribution event, in each case, plus any declared but unpaid dividends thereon; |
59
| ● | the Amended Articles divides the Holdco Board into three classes, each of which is elected once every three years with a Board of at least three and no more than 11 members; |
| ● | an amendment to the Amended Articles generally require, in addition to the approval of the Holdco Board of Directors, a vote of the holders of a majority of Holdco’s outstanding Holdco Ordinary Shares entitled to vote present and voting (including via proxy) on the matter at a general meeting of shareholders (referred to as simple majority), and the amendment of a limited number of provisions, such as the provision empowering the Holdco Board to determine the size of the Holdco Board, the provision dividing Holdco’s directors into three classes, the provision that sets forth the procedures and the requirements that must be met in order for a shareholder to require Holdco to include a matter on the agenda for a general meeting of the shareholders, and the provisions relating to the election and removal of members of the Holdco Board and empowering the Holdco Board to fill vacancies on the Holdco Board, requires, in addition to the approval of the Holdco Board, a vote of the holders of 65% of Holdco’s outstanding Holdco Ordinary Shares entitled to vote at a general meeting; in other cases, such as an amendment of the provisions relating to the rights and privileges of the Holdco Preferred Shares, the amendment of a limited number of provisions also requires the approval of the Preferred Majority; and certain amendments to the Amended Articles will not be applicable against any holder of Holdco Preferred Shares that does not consent to such amendment; |
| ● | the Amended Articles do not permit a director to be removed by a vote of the Holdco shareholders, without the approval of at least 65% of our outstanding shares entitled to vote at a general meeting of shareholders; and |
| ● | the Amended Articles provide that director vacancies may be filled by the Holdco Board. |
Further, Israeli tax considerations may make potential transactions undesirable to Holdco or to some of Holdco’s shareholders whose country of residence does not have a tax treaty with Israel granting tax relief to such shareholders from Israeli tax. For example, Israeli tax law does not recognize tax-free share exchanges to the same extent as U.S. tax law. With respect to mergers, Israeli tax law allows for tax deferral in certain circumstances but makes the deferral contingent on the fulfillment of numerous conditions, including a holding period of two years from the date of the transaction during which certain sales and dispositions of shares of the participating companies are restricted. Moreover, with respect to certain share swap transactions, the tax deferral is limited in time, and when such time expires, the tax becomes payable even if no disposition of the shares has occurred.
Holdco’s Amended Articles designate specific courts as the exclusive forum for certain litigation that may be initiated by its shareholders, which could limit such shareholders’ abilities to obtain a favorable judicial forum for disputes with Holdco or its directors, officers or employees.
Holdco’s Amended Articles provide that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act (the “Federal Forum Provision”). Investors cannot waive compliance with U.S. federal securities law and the rules and regulations thereunder. Holdco’s Amended Articles also provide that unless Holdco consents in writing to the selection of an alternative forum, the competent courts in Tel Aviv, Israel shall be the exclusive forum for any derivative action or proceeding brought on behalf of Holdco, any action asserting a breach of a fiduciary duty owed by any of its directors, officers or other employees to Holdco or its shareholders or any action asserting a claim arising pursuant to any provision of the Amended Articles, the Companies Law or the Israeli Securities Law (the “Israeli Forum Provision”).
The Federal Forum Provision and Israeli Forum Provision may limit a shareholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with Holdco or its directors, officers or other employees, which may discourage lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find the Federal Forum Provision or the Israeli Forum Provision to be inapplicable or unenforceable in an action, Holdco may incur additional costs associated with resolving such action in other jurisdictions, which could harm its business, financial condition or results of operations.
60
It may be difficult to enforce a U.S. judgment against Holdco, its officers, and its directors named in this Annual Reportin Israel or the United States, or to assert U.S. securities law claims in Israel or serve process on Holdco’s officers and directors.
Not all of the individuals that are Holdco’s directors or officers are residents of the United States, and most of their assets are located outside the United States. Service of process upon Holdco or its non-U.S. resident directors and officers and enforcement of judgments obtained in the United States against Holdco or its non-U.S. resident directors and officers may be difficult to obtain within the United States. We have been informed that it may be difficult to assert claims under U.S. securities law in original actions instituted in Israel or obtain a judgment based on the civil liability provisions of U.S. federal securities law. Israeli courts may refuse to hear a claim based on a violation of U.S. securities law against Holdco or its non-U.S. officers and directors because Israel may not be the most appropriate forum in which to bring such a claim. In addition, even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by Israeli law. There is little binding case law in Israel addressing the matters described above. Israeli courts might not enforce judgments rendered outside Israel, which may make it difficult to collect on judgments rendered against Holdco or its non-U.S. officers and directors.
Moreover, an Israeli court will not enforce a non-Israeli judgment if it was given in a state whose laws do not provide for the enforcement of judgments of Israeli courts (subject to exceptional cases), if its enforcement is likely to prejudice the sovereignty or security of the State of Israel, if it was obtained by fraud or in the absence of due process, if it is at variance with another valid judgment that was given in the same matter between the same parties, or if a suit in the same matter between the same parties was pending before a court or tribunal in Israel at the time the foreign action was brought.
Your rights and responsibilities as a Holdco Shareholder are governed by Israeli law, which differs in some respects from the rights and responsibilities of shareholders of U.S. corporations.
Holdco is incorporated under Israeli law. The rights and responsibilities of holders of Holdco Ordinary Shares are governed by the Amended Articles and Israeli law, including the Companies Law. These rights and responsibilities differ in some respects from the rights and responsibilities of shareholders in typical U.S. corporations. In particular, pursuant to the Companies Law, each shareholder of an Israeli company has to act in good faith and in a customary manner in exercising his, her or its rights and fulfilling his, her or its obligations toward a company and other shareholders and to refrain from abusing his, her or its power in a company, including, among other things, in voting at the general meeting of shareholders, on amendments to a company’s articles of association, and with regard to increases in a company’s authorized share capital, mergers, and certain transactions requiring shareholders’ approval under the Companies Law. A shareholder also has a general duty to refrain from discriminating against other shareholders. In addition, a controlling shareholder of an Israeli company or a shareholder who knows that it possesses the power to determine the outcome of a shareholder vote or who has the power to appoint or prevent the appointment of a director or officer in a company, or has other powers toward a company has a duty of fairness toward that company. However, Israeli law does not define the substance of this duty of fairness. There is little case law available to assist in understanding the implications of these provisions that govern shareholder behavior, but these provisions may be interpreted to impose additional obligations and liabilities on our shareholders that are not typically imposed on shareholders of U.S. corporations.
61
| ITEM 4. | INFORMATION ON THE COMPANY |
4.A. History and Development of the Company
Holdco Nuvo Group D.G Ltd. a limited liability company, was incorporated under the laws of the State of Israel on July 20, 2023 for the sole purpose of effectuating the Business Combination as described in the Introductory Note to this Annual Report. Following and as a result of the Business Combination, the business of Holdco is conducted through Nuvo Group Ltd., its direct, wholly-owned subsidiary, which was incorporated in Israel on June 28, 2006.
Our principal executive offices are located at 94 Yigal Alon St., Tel Aviv, Israel 6789155 and our telephone number is: 1-800-554-9041. Nuvo’s wholly owned U.S. subsidiary, Nuvo Group USA, Inc., incorporated in Delaware, has been appointed our agent in the United States, and its registered address is 300 Witherspoon Street, Suite 201, Princeton, New Jersey 08542. Our website address is https://www.nuvocares.com. The information contained on, or that can be accessed through, our website is not part of this Annual Report on Form 20-F. We have included our website address herein solely as an inactive textual reference. The SEC also maintains a website that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC. The address of this website is http://www.sec.gov.
See “Operating and Financial Review and Prospects—Liquidity and Capital Resources” for a discussion of Nuvo’s capital expenditures.
4.B. Business Overview
Overview
Our Business
We believe Nuvo has the potential to become a leader in remote fetal monitoring for pregnancy care. We are leading the transformation from a world where pregnancy care is limited by outdated technology and barriers to accessing care to a world where data-driven, clinically relevant, actionable insights can be accessed both at home and in the clinic, during the 32nd week of pregnancy until the beginning of labor (the “INVU monitoring period”), by an expectant mother and her clinician. Current poor fetal and maternal health outcomes, limited accessibility to care, and soaring costs all indicate the need for a change in the way that pregnancies are monitored and managed, and we believe Nuvo’s innovative solution, which we refer to as our INVU platform, is the only solution that is positioned to address complete accessibility to care while looking significantly deeper into the pregnancy than standard of care solutions do today. Recognizing that the tools used today to monitor and manage pregnancies may not be the same tools used a decade from now, Nuvo believes its solution is well positioned to be at the forefront of this market shift. Strategically, Nuvo’s platform is currently being commercialized by tapping into a key part of the pregnancy journey, fetal non-stress tests (“NSTs”), by enabling these tests to be conducted remotely with clinical accuracy that has been demonstrated to be equivalent to the standard of care based off of our clinical studies and consumer-grade ease of use (see “—Clinical Studies”). NSTs are medically necessary pregnancy screening procedures that measure fetal heart rate and reaction to movement to assess fetal well-being. NSTs are most commonly conducted with cardiotocography (“CTG”) machines, which were designed for intrapartum monitoring in clinics by experienced healthcare professionals. Through a combination of advanced wearable technology, AI & machine learning, and compelling user experiences (for expectant mothers and clinicians), INVU by NuvoTM (“INVU”) enables increased access to care, deeper insights into maternal-fetal health, reduced clinical staff burden, and improved patient satisfaction.
62
For a remote fetal and maternal monitoring program to be successfully implemented, we believe that the monitoring device should do the following: (i) be designed for self-application by the expectant mother and without the need for device repositioning by a medical professional; (ii) acquire valid data that accurately distinguishes between maternal and fetal heart rate (“MHR” and “FHR”, respectively); (iii) be capable of continuously monitoring MHR and FHR during the times in a pregnancy when protocol requires monitoring; (iv) have a very low rate of false results, such as detecting a fetal heartbeat when there is none or inaccurately detecting heart rate, to prevent false reassurance or anxiety outside of a clinical environment; (v) be comfortable; and (vi) have the capability of measuring other variables such as maternal uterine activity (“MUA”), more commonly known as contractions, maternal and fetal electrocardiography, measuring the heart’s electrical activity and the pattern of the heart beats (“mECG” and “fECG”, respectively) and others to offer other tests, such as NSTs, and be able to analyze such data to identify phenomena and to develop screening and predictive models, including through the discovery of biomarkers, and enable population health strategies. However, currently available technology has difficulty reliably and efficiently measuring most of the above, has not proven to reliably measure FHR at certain times in a pregnancy or MUA in most cases, if at all, and does not aggregate and analyze data in a sophisticated manner. Our INVU platform was designed to be a fully remote, medical-grade maternal and fetal monitoring solution that addresses each of the aforementioned challenges and more.
INVU is composed of a hardware component (wearable), with digital signal processing and cloud analytics, and interfaces for every participant involved in the pregnancy care. The hardware component of our INVU platform is a proprietary self-administered wireless sensor band that clinicians prescribe to expectant mothers who wear the sensor band during virtual visits to capture real-time data on key maternal and fetal health metrics. During these visits, a live reading allows the expectant mother to access simplified data and insights via the paired INVU application. Our wireless sensor band captures a unique set of in-depth physiological data from the expectant mother and unborn baby in a passive manner, without sending energy signals into the womb. Next, the data is digitized and sent wirelessly for analysis on our cloud-based servers by our sophisticated algorithms. Today, when obstetrics clinicians connect to our INVU platform, they have access to a digital dashboard that contains fetal and maternal heart rate and uterine activity tracings recorded during the session and data derived from these measurements for all expectant mothers and unborn babies in their care that use our INVU platform. This data is comparable to the fetal surveillance procedures that normally occur once or twice weekly in the last trimester of pregnancies1 that have some indication for risk. According to a study in the American Journal of Obstetrics and Gynecology (“AJOG”) analyzing approximately ten million pregnancies, 38% were identified as low risk and 62% were identified as high risk for unexpected complications.2
Our INVU platform is also capable of integrating with other peripheral and medical devices, such as blood pressure cuffs, subject to and in accordance with FDA regulation, which would allow expectant mothers and their clinicians to easily record and track important vitals all on one application to inform personalized care plans. In the future, we intend to seek FDA clearance to use advanced machine-learning and AI capabilities to analyze the data we collect to provide clinicians and expectant mothers with significantly more actionable predictive data and insights. In order to do so, we have developed an external data platform which automatically captures and analyzes all data recorded by our INVU platform in research, clinical and commercial domains, to the extent we have a data sharing agreement in place, which we believe will enable the rapid development of future AI models. First, we plan to provide a rule-based decision support system based on the automation of existing clinical guidelines to support clinicians in clinical decisions they are already making, which we believe will only require technical validation. Second, we plan to develop AI models aimed at providing obstetrics clinicians with new information they otherwise would not have access to, such as predicting risks before they become visible later in pregnancy, which will require clinical validation and FDA clearance. However, there is no assurance that we will be able to develop such rule based decision support system or AI models as planned or if developed, that such programs will be received favorably by clinicians or expectant mothers.
Currently, our products are categorized as Class II devices and subject to the premarket notification requirements under section 510(k) of the Federal Food, Drug, and Cosmetic Act of 1938 (the “FDCA”). Our INVU platform received 510(k) clearance from the FDA in March 2020 to conduct a five-minute trace of MHR and FHR, for singleton pregnancies, or a pregnancy with one baby, from the 32nd week of pregnancy until the beginning of labor. We refer to this five-minute trace as a fetal surveillance and to this time frame as the INVU monitoring period. MUA, more commonly known as contractions, and its intended use, in conjunction with MHR and FHR, for NSTs during the INVU monitoring period, received FDA clearance in May 2021, allowing us to perform fetal surveillance and measure MUA, and as a result, offer NSTs during the INVU monitoring period.
| 1 | The American College of Obstetricians and Gynecologists, “Indications for Outpatient Antenatal Fetal Surveillance”, ACOG Committee Opinion, Committee on Obstetric Practice Society for Maternal-Fetal Medicine, Volume 137, Number 828 (June 2021). | |
| 2 | American Journal of Obstetrics and Gynecology, “Unexpected complications of low-risk pregnancies in the United States”, Volume 212, Issue 6, Article (June 2015). |
63
The 510(k) process and de novo classification are two regulatory pathways for medical devices in the United States. The 510(k) is used for devices that are “substantially equivalent” to existing ones (predicate devices), with a shorter review time than a device that would need a pre-market approval (“PMA”). De novo pathway is for novel or low-risk devices that lack comparable predicates and may require more data and a longer review. The 510(k) classifies devices into Class I, II, or III based on risk, while de novo can create a new classification. See “—Government Regulation.”
Nuvo has received 510(k) clearance for INVU. Specifically, the Indication for Use describes:
| ● | INVU is a maternal-fetal monitor that non-invasively measures and displays FHR, MHR and MUA. |
| ● | The INVU sensor band acquires the fetal heart electrocardiogram and maternal heart electrocardiogram signals from abdominal surface electrodes and the fetal phonocardiogram and the maternal phonocardiogram signals from surface acoustic sensors. The FHR, MHR and MUA tracings are derived from these signals and presented. |
| ● | INVU is indicated for use by pregnant women who are in their 32nd week of gestation (or later), with a singleton pregnancy. |
| ● | The INVU maternal-fetal monitor is intended for use by healthcare professionals in health care facilities and by the patient in the patient’s home, on the order of a physician. |
| ● | INVU is indicated for antepartum fetal surveillance (i.e. NSTs). |
In addition to the FDA clearances mentioned above, we also received ISO 13485 certification in February 2020 for the development, manufacturing, marketing and sales of pregnancy monitoring devices. Additionally, in November 2020 we received certification from the Medical Device Division, or AMAR, of the Ministry of Health in Israel, which grants the ability to commercialize our INVU technology in Israel. This certification is valid through May 2024 and the approval and indications for use are in accordance with the FDA clearance mentioned above. Renewal time for the certification can take up to 120 days and if there are no critical changes to the device, then the expectation is that we would receive an extension to the certification. We have not obtained additional regulatory approval of our INVU platform or any of our other products outside of the United States and Israel and have not taken steps to do so, other than with respect to filing for a CE mark in Europe in March 2023 to offer NSTs using our FHR, MHR and MUA capabilities. Approval or clearance from the FDA, or comparable regulatory agency in other jurisdictions, to capture certain measurements and perform certain tests using our INVU platform, is not guaranteed and may take longer than planned. Also, regulatory approval in one jurisdiction does not mean that we will succeed in obtaining regulatory approval in other jurisdictions. We are commercializing our INVU platform with an initial focus on large healthcare systems and obstetrician-physician practice management groups. In addition, obstetrician-physician practice management groups play a significant role in pregnancy care management during the INVU monitoring period, as they work with expectant mothers starting early in the pregnancy and therefore have a strong interest in improving pregnancy care during the period of pregnancy before labor, or antepartum period. We believe these groups will be most effective at implementing our technology in clinical practice due to the continuous flow of expectant mothers that they care for and their desire for improved outcomes at reduced cost that we believe our INVU platform will ultimately enable. We believe our INVU platform is the first FDA-cleared device to measure FHR, MHR and MUA passively, remotely and with a self-administered wireless sensor band that utilizes direct physiological signals during the antepartum period for NSTs. Our technology acquires and records deep and rich data outputs during each monitoring session through our biopotential sensors, that detect electrical signals, and acoustic sensors, and extracts multiple physiological measurements from these data, including mECG and fECG, and maternal and fetal phonocardiography (“mPCG” and “fPCG”, respectively). The mPCG signal reflects the activity of the maternal heart while fPCG is a method based on sensing the acoustic signals of the fetal heart from the maternal abdomen. We believe that these and other metrics based on our collected data will be critical to pregnancy management tests and procedures we plan to offer in the future. Over the longer term, we intend to conduct clinical trials to examine the impact of our INVU platform on monitoring compliance, quality of care and healthcare outcomes, as well as costs.
64
We have over a dozen commercial agreements, including purchase orders, with health systems, large private practice groups and independent women’s health practices in the United States and Israel. We refer to our commercial contracts with major healthcare systems and obstetrician-physician practice management groups as enterprise level agreements. For a discussion of our current and intended enterprise level agreements, please see “—Our Revenue Model.” If we demonstrate that our INVU platform increases monitoring compliance, improves quality of care and healthcare outcomes, as well as reduces payer costs, we expect to focus on seeking long-term contracts with payers that allow us to benefit from a percentage of any cost-savings that we achieve. We also believe that any cost-savings achieved from utilizing our INVU platform will incentivize payers to encourage their obstetrician networks and expectant mothers to utilize our INVU platform.
We intend to apply data algorithms and other innovative digital tools to conduct AI-powered machine learning and computer-based predictive analytics to make targeted predictive recommendations to individual expectant mothers who have health profiles for which we have identified particular, notable patterns and trends. We believe these predictive insights, such as identifying risks before they become visible later in pregnancy, will help clinicians to improve monitoring schedules and frequency and identify appropriate times to intervene for individual pregnancies and facilitate population health strategies aimed at improving a specific population’s health outcome as efficiencies are improved and costs are reduced. We expect that our ability to develop biomarkers and predictive analytics will set us apart from other pregnancy management monitoring systems and make us more effective at enabling proactive pregnancy management to improve outcomes for expectant mothers and unborn babies.
Our innovative technology is protected by an extensive global patent portfolio consisting of 16 issued U.S. patents, 10 pending U.S. utility patent applications, 43 issued or allowed foreign patents, 13 pending foreign utility patent applications and one pending international (“PCT”) patent application, which we constantly review and seek to expand. We believe that we will be able to obtain patents relating to data input, the means of analysis and the output from such analysis. We believe that our technology and the protection that we have afforded it currently give us a significant competitive advantage and is a barrier to competitors. Subject to the receipt of required regulatory clearances and approvals, we expect to further strengthen our INVU platform by gathering and analyzing more data and potentially identifying patterns and trends to develop predictive models and population health strategies.
65
Our Platform
Our INVU platform was designed to allow for flexibility in implementation. In our current commercial model, the expectant mothers access prenatal care at home according to their clinician’s protocol, through a self-administered and easy to use wireless sensor band that connects to our cloud-based platform and provides personalized clinical-quality care in a virtual environment, in real time.
The prescription initiated, protocol-driven process from the expectant mother’s completion of monitoring to return of our device is demonstrated below.
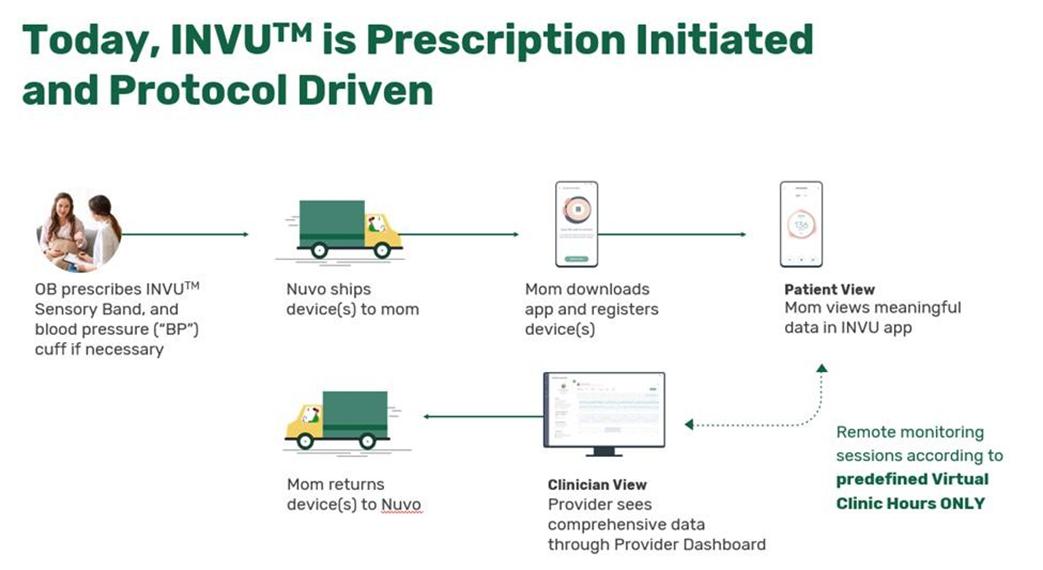
The “collect, compute and visualization” process of our wireless sensor band is demonstrated below.
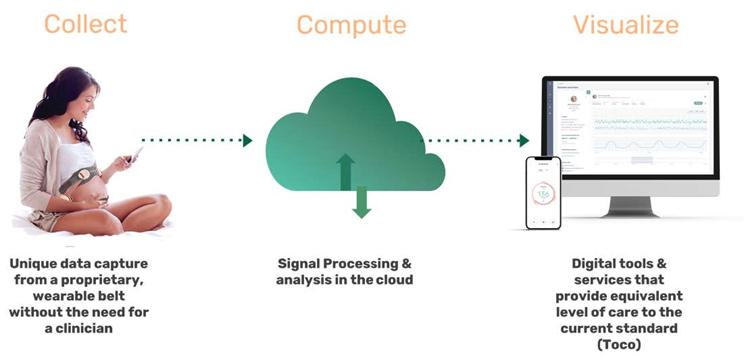
66
Key Attributes
We believe our INVU platform provides or will provide the following key benefits for expectant mothers, unborn babies, clinicians and payers. While some of the following benefits are benefits currently being provided with our FDA-cleared solution, others represent our goals with respect to the INVU platform that will require more data and monitoring and we may ultimately not be able to provide such benefits.
| ● | Increased access to care: Expectant mothers can access clinical-quality pregnancy care anytime, anywhere according to clinician protocol, during the antepartum period subject to any restrictions as to time and place in any FDA clearance, without the need to travel to medical offices or spend time in waiting rooms and regardless of clinician proximity. We believe, according to Nuvo estimates and information provided by the American College of Obstetricians and Gynecologists (“ACOG”)3, that the INVU platform can save expectant mothers 14 half-days of paid time off, 32 hours of travel time and approximately $300 in travel expenses, which we believe can lead to a 15-20% increase in workplace productivity amongst expectant employees. This is a benefit currently being provided with our FDA-cleared solution. |
| ● | Improved user experience: Expectant mothers can administer our wireless sensor band without assistance from a medical professional. Clinicians can integrate our INVU platform with other existing systems and protocols, subject to FDA clearance in some cases, can easily schedule monitoring sessions and conduct monitoring on short notice on a near real time basis if concerns arise, and can send messages to the expectant mothers in their care. Expectant mothers and their clinicians are reassured through the connection of the expectant mother to her care team and the near real-time, medical-grade data on key pregnancy health metrics that they each receive. This is a benefit currently being provided with our FDA-cleared solution. |
| ● | Reduced cost of care: We believe that use of our INVU platform will lead to fewer required in-person visits by expectant mothers to clinicians and healthcare facilities, and ultimately fewer procedures, which would result in lower costs across the healthcare system. We believe, according to Nuvo estimates, using datapoints from the US Centers for Disease Control and Prevention (“CDC”) relating to the number of annual pregnancies in the United States4, the average cost of emergency department visits from a 2019 United Health Group report5 and information from a study contained in the AJOG in 20176, that OB-ED avoidance alone in the United States would represent approximately $2.4 billion in system cost savings. Additionally, we believe, according to Nuvo estimates, using datapoints from Zipia regarding average OBGYN nurse salary7 and MDSave regarding OBGYN estimated average costs for patient office visits8, that approximately $21,000 would be saved in nurse time per year and the incremental annual revenue potential could be approximately $475,000. This is a benefit that may be provided with our FDA-cleared solution, but will require more data and monitoring of that data to support this claim definitively. In addition, future products, subject to FDA approvals, may further Nuvo’s claims on this topic. |
| ● | Improved outcomes: We believe that expectant mothers will be more likely to comply with our monitoring protocols, which, together with other benefits of our INVU platform, has the potential to result in better health outcomes if the frequency of complications and other events, such as C-sections, emergency department visits, hospital stays and neonatal intensive care unit stays are reduced. We intend to use integrated data and proprietary predictive analytics to develop personalized care recommendations for expectant mothers. This is a benefit that may be provided with our FDA-cleared solution, but will require more data and monitoring of that data to support this claim definitively. In addition, future products, subject to FDA approvals, may further Nuvo’s claims on this topic. |
| 3 | The American College of Obstetricians and Gynecologists, “Indications for Outpatient Antenatal Fetal Surveillance”, ACOG Committee Opinion, Committee on Obstetric Practice Society for Maternal-Fetal Medicine, Volume 137, Number 828 (June 2021). | |
| 4 | US Centers for Disease Control and Prevention, National Vital Statistics Reports, “Births: Final Data for 2021”, Volume 72, Number 1 (January 31, 2023). | |
| 5 | United Health Group, “18 Million Avoidable Hospital Emergency Department Visits Add $32 Billion in Costs to the Health Care System Each Year”, Report (July 2019). | |
| 6 | American Journal of Obstetrics and Gynecology, “Non-Urgent and Urgent Emergency Department Use During Pregnancy: An Observational Study”, Volume 216, Issue 2 (February 2017). | |
| 7 | Zippia, “OB/GYN Nurse Salary”, Article (Updated September 14, 2023). | |
| 8 | MDSave, “OBGYN Established Patient Office Visit”, Article (Updated 2024). |
67
| ● | Improved population health strategies: We believe that our future ability to analyze aggregated data will enable us to make highly useful and actionable predictive recommendations which will result in a healthier population of expectant mothers and unborn babies. This is a benefit that may be provided with our FDA-cleared solution, but will require more data and monitoring of that data to support this claim definitively. In addition, future products, subject to FDA approvals, may further Nuvo’s claims on this topic. |
We believe our INVU platform is the only platform that contains all of the above attributes and that also (i) utilizes multimodality technology in one instrument to monitor pregnancy, (ii) utilizes electrocardiography and phonocardiography for remote monitoring, (iii) can monitor continuously, passively and remotely in accordance with clinician-prescribed protocol when the expectant mother is wearing our wireless sensor band, (iv) provides substantially equivalent results to CTG, which is the existing standard of care for pregnancy care monitoring and offers NSTs passively, remotely and through self-administration during the INVU monitoring period, (v) delivers high resolution and personalized medical-grade data to the clinician and the expectant mother and (vi) has the potential to aggregate data and apply innovative digital tools to make targeted predictive recommendations, as well as enable population health strategies.
Expectant Mother Experience
Upon receiving our wireless sensor band, an expectant mother is provided detailed instructions for using and wearing the device, and utilizing the INVU application to conduct a session with her clinician. During the session, the wireless sensor band collects data that is transmitted to the expectant mother’s mobile device, and then from the mobile device to the cloud and from the cloud to the expectant mother and clinician in the form of meaningful outputs. In effect, the wireless sensor band serves as the bridge to connect the expectant mother and clinician to relevant data. An example of the interface that an expectant mother could see during a session is below:
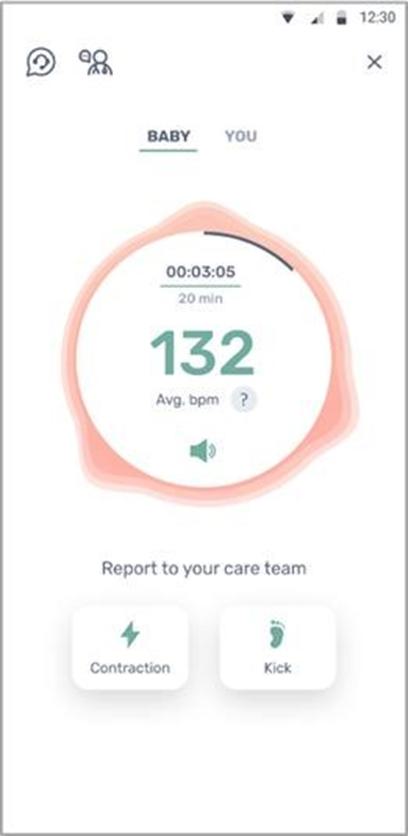
68
During a monitoring session, the expectant mother uses the INVU mobile app interface shown above to view her and her unborn baby’s average heart rates, and self-report kicks and contractions. The expectant mother can also use the app interface to communicate with Nuvo customer support as well as respond to messages sent by her care team.
Currently, we have received regulatory clearance for measuring FHR, MHR and MUA, and as a result, offering NSTs, during the INVU monitoring period for a singleton pregnancy. Although the sensor band will be recording significantly more data than that, and will be recording beat-by-beat MHR and FHR, the expectant mother only receives an average MHR and FHR measurement, as well as any fetal kicks and contractions that she separately logs. At the end of the session, an expectant mother will receive a session summary, including the average MHR and FHR.
Obstetrics Clinician Experience
We believe our INVU platform will lead to improved throughput, as Nuvo’s average monitoring time for NSTs is 28.9 minutes, inclusive of staff time. In addition, those utilizing our INVU platform are able to run multiple NSTs simultaneously, creating an estimated 60 minutes of freed exam room time per NST that can be used towards other billable procedures, plus the savings in clinical staff time.
Clinicians have access to an application known as INVU Pro that provides a digital dashboard reflecting all expectant mothers under their care. This dashboard may be sorted into different presentations and modified by the clinician based on their preferences. The clinician enters details about the expectant mother and pregnancy and establishes a protocol or care plan on the application, which includes the time, frequency and duration of the monitoring sessions. Care plans may also call for blood pressure monitoring before or after the sessions. The clinician may change the protocol or care plan in the application. Sessions may only be prescribed by the clinician and conducted during scheduled times. Our wireless sensor band cannot be used to record any data at other times. The clinician may view the session live or after it is completed. An example of a screen a clinician might see is below.
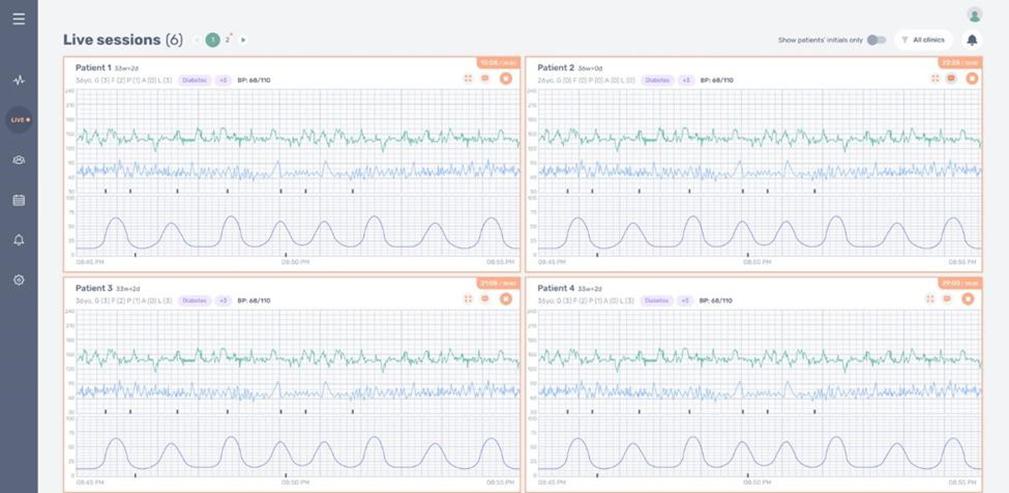
The live sessions screen in INVU Pro displays up to four simultaneous monitoring sessions on each page. Each monitoring session displays the expectant mother’s clinical details and NST data, which includes the FHR in green, MHR in blue, and the MUA. The clinician can double-click on any of the four sessions to see an expanded view with more detail and the expectant mother chat interface.
At the end of the session, the expectant mother will receive a session summary, including the average MHR and FHR, and the clinician will receive more detailed information relating to MHR and FHR based on the clinician’s stated preference. The application allows a clinician to write notes to the expectant mother. This feature is useful for conveying information to the expectant mother about her pregnancy, the unborn baby or changes in protocol.
69
Future Plans and Expectations
We intend to seek clearance to extend our INVU monitoring period as well as report other measurements. Approval or clearance from the FDA, or comparable regulatory agency in other jurisdictions, to capture certain measurements and perform certain tests, is not guaranteed and may take longer than planned. If we obtain additional clearances to report other measurements that our INVU platform is able to capture, compute and visualize, we will be able to provide and market additional pregnancy health metrics to participants in the pregnancy care management process, including expectant mothers and clinicians. We intend to utilize the data we collect, combined with external guidelines, to establish cloud-based decision support systems. We also expect to add external data provided by third-party sources and harmonize all data we have collected into one cohesive set. We intend to develop decision support tools to analyze the data we collect to develop and execute new personalized care protocols and population health strategies which we believe will enhance value-based care models. We also intend to apply data algorithms and other innovative digital tools to conduct AI-powered machine learning computer analyses to identify patterns and trends based on the data and to develop predictive models to ultimately enable population health strategies.
In addition to providing better care for expectant mothers and unborn babies, we believe that as our INVU platform develops, clinicians will be able to use existing and expanded current procedural terminology (“CPT”) codes, which offer clinicians a uniform language for coding medical services and procedures, for reimbursement for expectant mothers utilizing our platform and to employ our INVU platform, to maintain or increase their revenue and improve effectiveness. This is due, in part, to the additional efficiencies which we believe will allow them to increase the number of expectant mothers they care for and be more available when procedures are required. See “—Reimbursement and Payment.”
To convince more clinicians, we will need to present sufficient impact evidence of the increased monitoring compliance, improved quality of care and healthcare outcomes, as well as reduced payer costs, obtained from our services to our commercial customers. If we can provide such evidence, we believe our commercial customers will be more likely to incentivize payers to enter into value contracts and partner with them, and payers will be more likely to encourage their obstetrics clinician networks and expectant mothers to utilize our services.
As part of our operational strategy, we have established a third-party servicing center where our wireless sensor bands are prepared for use by the next expectant mother after thorough cleaning and quality control testing and, if needed, fixing and refurbishing, which could involve replacement of some sensors. See “—Manufacturing and Supply.” We believe that, on average, a wireless sensor band should be viable for approximately 12 mothers over a three-year period before needing to be replaced.
Our Revenue Model
Current State
We have begun to fully commercialize the INVU platform and have signed over a dozen commercial contracts with health systems, large private practice groups and independent women’s health practices in the United States and Israel. Nuvo utilizes the net promoter score (“NPS”) customer satisfaction methodology that is based on a single survey question asking respondents to rate the likelihood that they would recommend a company, product, or a service to a friend or colleague. Promoters are customers who respond with a score of 9 or 10. Passives are customers who respond with a score of 7 or 8. Detractors are customers who respond with a score of 0 to 6. The NPS score is calculated with the following formula: total % of promoters – total % of detractors = NPS. We surveyed healthcare providers and expectant mothers at initial onboarding, after INVU monitoring sessions, and following the end of their care plans. As of May 2024, the reported NPS for expectant mothers was 68 and 45 for clinicians, on a scale of -100 to 100. These scores exceed overall NPS and healthcare industry averages, according to Survicate’s 2022 NPS Benchmark Report9.
We are currently focused on the most significant providers of prenatal care in the United States that often control the entire pregnancy journey. Our primary initial customer focus is on long-term enterprise level agreements with larger obstetrician-physician practice management groups, large hospital networks and U.S. healthcare systems, that we believe will be most effective at implementing our technology into clinical practice, with weighted emphasis on systems that provide pregnancy care from the beginning of the pregnancy, are leading providers of antenatal care and are stronger in value-based contracting with payers, particularly in connection with high-risk pregnancies (“HRPs”). We are structuring these agreements to include up-front and ongoing platform payments. Initial agreements may provide for introductory preferential pricing as an incentive to try the platform, and we are examining a number of other models, including fee-for-service, volume pricing or device purchasing.
| 9 | Survicate, “NPS Benchmarks for 2023: Good Net Promoter Scores by Industry”, NPS Benchmarks Report (March 31, 2023). |
70
Future State
If we establish evidence of the short and long-term benefits of our INVU platform and our ability to generate value for payers, we aim to seek long-term contracts with payers, primarily insurers and self-insured employers. Under these agreements, while we will typically initially receive payments as providers are paid, we ultimately expect to receive payments for the use of our INVU platform based, at least in part, on a percentage of cost-savings achieved by the applicable payers, which we believe is potentially larger. The expectation of reduced costs has been supported by at least one study in Denmark10 that demonstrated that an at home pregnancy solution involving nurses bringing hospital equipment to the home of a mother led to a 44% reduction in bed occupation for HRPs, 75% less time spent on patient monitoring by hospital staff and 93% improved patient satisfaction. Although not measured directly in this study, the reductions in bed occupation and time spent on patient monitoring would be expected to reduce costs associated with pregnancy care. We believe that our data tools and actionable insights we may develop, for example, through increased monitoring compliance and refined care protocols based upon our proprietary data analyses, should allow payers to realize significant cost-savings through the implementation of INVU in their population, and that if we are able to demonstrate improved quality of care and healthcare outcomes and cost savings, payers should be incentivized to encourage their obstetrics clinician networks and expectant mothers to utilize our INVU platform.
We believe that the data we expect to acquire from expectant mothers and unborn babies we monitor as we expand and improve our pregnancy care platform, the clinical innovation that we expect to result from the decision support tools we expect to develop and the insights we expect to gain and predictive models created based on our analysis of such data with these tools, will lead to increased efficiency and improved outcomes. We intend to monetize these capabilities, if developed, by selling to payers the anonymized population insights we obtain through our INVU platform, and by sharing in the cost savings that payers realize from these insights. We believe that payers will have a strong interest in what we believe will be high quality, aggregated pregnancy care data, the analysis of such data, actionable insights for the expectant mothers in their systems and to assist in managing population health, in large part to optimize payer spending, as they have with other specialties where remote monitoring platforms have become popular. As broader adoption occurs, we expect to generate greater enterprise level agreement revenues, as well as greater monetization potential as our data pool grows.
Our Competitive Advantages
We believe the following combination of strengths, capabilities and features of our data-driven connected pregnancy care management platform distinguishes us from our competitors and positions us to successfully compete, to address certain market opportunities and weaknesses, and to disrupt the pregnancy care management and monitoring market, through our innovative INVU platform. However, see also a discussion of the various risks we face under “Risk Factors—Risks Related to Our Business and Our INVU Platform.” We also believe that the pregnancy care management and monitoring market will show significant growth over the next years and decades, and that we are well-positioned to benefit from such growth.
Our INVU platform increases access to care through a remote solution. In recent years, telehealth infrastructure and remote monitoring solutions have become indispensable tools for care delivery and medical practice revenue generation. Pregnancy care has lagged behind other medical fields with respect to remote care due to difficulty in developing remote technology that can be easily administered early in the pregnancy without assistance and that provides monitoring within protocols at home that yield substantially equivalent results as the current standard of care obtained within healthcare facilities. Our INVU platform, which we believe is the only platform that uses new technology cleared by the FDA for use during the antepartum period, will be of particular benefit to three types of expectant mothers. First, those living in rural areas without access to care, or those whose clinics see a high volume of expectant mothers. According to the March of Dimes11, as of 2022, there were approximately seven million women in the United States living in counties without access or with limited access to maternity care, giving birth to more than 500,000 babies a year. Second, expectant mothers with lower risk pregnancies, or LRPs, can enjoy the freedom associated with remote visits resulting in fewer in-office visits, easier compliance with care protocols and access to virtual triaging. Third, expectant mothers with HRPs who can also travel less frequently for in-office visits should enjoy more flexibility and freedom in receiving the proper level of care that they need, both at home and in the clinic during the INVU monitoring period, which we believe will result in a higher likelihood of monitoring compliance and reassurance for the expectant mother. According to the University of California, San Francisco Health12, high-risk complications occur in 6% to 8% of all pregnancies in the United States. In addition, COVID-19 and variants of the virus further accelerated acceptance of remote preventive care by both care providers and patients. We believe our INVU platform will be unmatched in remote care and based on the results of our clinical studies, in distributing access to enhanced pregnancy care.
| 10 | Acta Obstetricia et Gynecologica Scandinavica, “Home management by remote self-monitoring in intermediate- and high-risk pregnancies: A retrospective study of 400 consecutive women”, Volume 101, Issue 1, Article (January 2022). |
| 11 | March of Dimes, “Nowhere to Go: Maternity Care Deserts Across the U.S.”, Maternity Care Deserts 2022 Report. |
| 12 | University of California, San Francisco Health, “Obstetrics & Gynecology: High-risk pregnancy”, Article (Updated 2024). |
71
Our wireless sensor band is designed to be self-administered by expectant mothers both at home and in the clinic. Our wireless sensor band was designed from the perspective of the expectant mother to be easy to use and applied by the expectant mother after reviewing simple virtual instructions, beginning in the 32nd week of pregnancy throughout the pregnancy journey, both at home and in the clinic. Most other devices that are labeled as remote-use devices are, in effect, miniaturized CTGs, whether using Doppler ultrasound, a tocodynamometer (“TOCO”), a device used to measure the length, frequency and strength of uterine contractions, or both, which have proven to be difficult to properly self-administer without assistance from a medical professional, especially for prolonged procedures such as NSTs. We believe an expectant mother’s ability to utilize our INVU platform both at home and in the clinic, and without assistance, significantly enhances the expectant mother’s experience, and we expect will increase compliance with routine monitoring protocols.
Our INVU platform has the potential to enable proactive pregnancy management, which we believe will result in better outcomes. Today, our INVU platform is capable of helping clinicians provide expectant mothers with routine prenatal care and offer NSTs, in each case during the INVU monitoring period for singleton pregnancies. As we offer more NSTs, we will capture more data in a less costly manner, which we believe, among other things, will help to identify patterns and trends that may allow detection of certain complications earlier and facilitate timely intervention. Our INVU platform provides both expectant mothers and clinicians with comparable and, potentially better, information than they would receive at a doctor’s office or a hospital. As we continue to utilize our measurement capabilities, we expect to develop biomarkers and predictive analytics, which we believe will set us apart from our competitors and make us more effective at enabling proactive pregnancy management to improve outcomes for expectant mothers and unborn babies.
Our INVU platform should make clinical-quality remote pregnancy care more attractive for clinicians and payers, which we believe will increase adoption as a new standard of care. We believe clinicians will be incentivized to prescribe our solution because they will gain access to medical-grade data remotely, efficiently and with less effort for both expectant mother and clinician. These features are tangible differentiators in the practitioners’ service offerings, which we expect to create the opportunity to strengthen the relationship to the expectant mother via virtual visits, while also potentially enabling increased compliance and better outcomes. Ultimately, the ability for improved virtual access to the expectant mothers they care for may allow clinicians to reallocate scarce resources to cases where their physical presence and care is otherwise required. We believe payers will be incentivized to adopt our solution as a way to rationalize systemic healthcare costs. Keeping expectant mothers out of high-cost settings, such as hospitals and doctors’ offices, except when absolutely necessary, should reduce costs to payers and self-insured expectant mothers. In addition, increased compliance with monitoring protocols has the potential to allow clinicians to identify certain complications earlier and possibly prevent expensive procedures. Furthermore, certain NSTs and potentially other procedures in the future, which are part of HRP management regimens, will no longer need to be performed in an office or hospital during the INVU monitoring period. We believe that the ability to perform these tests remotely should further reduce costs to payers as NSTs are frequently used in the management of HRPs, among other things. Even expectant mothers with LRPs should benefit through their improved access to basic prenatal care and virtual triaging and visits, as well as on transportation, childcare, and missed work costs, among other costs. We believe we will be able to provide impact evidence to clinicians and payers of the cost-benefits of our solution.
Our distributed care technology provides detailed data to enable population health strategies, and our database becomes harder to replicate as it grows. Through our advanced, multi-modality pregnancy monitoring technology, we believe we capture more detailed and granular signals and multiple physiological measurements, remotely, passively and in near real time, and acquire significantly more data from the expectant mother and unborn baby than other pregnancy monitoring systems. As we validate, aggregate and analyze data with respect to other physiological measurements, such as fECG, mECG, fPCG, mPCG, fetal activity, fetal position, maternal respiration and other measurements, we will strengthen our predictive abilities and, we believe, will eventually be successful in tackling significant pregnancy challenges, such as C-sections, preeclampsia, fetal and maternal arrhythmia and mood disorders. As our database increases with additional expectant mothers being monitored and data from clinical studies and other third-party sources, we plan to aggregate data in the cloud and combine it with existing guidelines to develop decision support systems, harmonize our data into one cohesive set, and apply data algorithms and other innovative digital tools to conduct AI-powered machine learning and computer-based predictive analytics. In addition, we expect these results will enable us to develop personalized and predictive care pathways for expectant mothers and unborn babies, make Al-based recommendations for treatment to the clinician, and provide more personalized care and better outcomes for expectant mothers and unborn babies. In addition, we believe these results will enable us to develop population health strategies to tackle significant pregnancy challenges. As a result, if we are able to establish and develop such capabilities, we believe that we will be able to increase our prospective revenues, make it harder for competitors to replicate our capabilities and establish INVU as the standard for remote pregnancy care management.
72
We have a comprehensive intellectual property portfolio. Our innovative technology is protected by an extensive global patent portfolio consisting of 16 issued U.S. utility patents, 10 pending U.S. utility patent applications, 43 issued foreign utility patents, one allowed foreign utility patent application, 13 pending foreign utility patent applications and one PCT patent application. Our patent portfolio also includes three issued U.S. design patents and seven issued foreign design patents. Our patents cover various aspects of our INVU platform, such as numerous remote, non-invasive techniques for monitoring vital signs such as MUA and fetal cardiac activity. Our patents also cover techniques for the use and analysis of ECG and acoustic signal data for various purposes, including the generation of various signals. Our patents also cover hardware elements of our INVU platform, including sensor-laden garments, acoustic sensors and electrodes. We have protected our intellectual property rights through our patent portfolio and have maintained and executed on deliberate innovation areas to sustain the continued growth of our patent portfolio. In addition, we own trade secrets and research and development know-how supporting our INVU platform. Our comprehensive portfolio of intellectual property enables our highly advanced INVU platform, and we believe it would be difficult for a competitor to develop an equivalent product without considerable time and expense.
Our senior management team and Board have deep industry experience. Our organization is characterized by a strong, entrepreneurial corporate culture that fosters our vision of improved, remote, accessible and affordable pregnancy management. Our senior management team and Board consist of seasoned medical device and other professionals, with a wide array of experience, including women’ health, medical technology, medical or healthcare, data science, marketing, financial, consumer products, clinical, navigating regulatory pathways, manufacturing, human resources and commercial expertise. Our Board has significant and diverse public market expertise in small and large U.S.-listed companies, as well as executive leadership experience in listed digital healthcare companies. Together, we have over a century of experience in operating, growing and overseeing multi-national companies and healthcare related businesses. Our experience spans from building and scaling consumer facing medical products and digital healthcare solutions, to leading large U.S.-based hospital groups and running global multi-billion dollar revenue companies. We believe our mission-driven team spirit, diverse background and significant experience in our industry, positions us to excel and deliver against our strategic objectives.
Our Growth Strategies
Our goal is to become the standard of care for remote pregnancy monitoring and pregnancy care management through the development of our INVU platform. To achieve our growth plan, we expect to employ the following core strategies. Such strategies may be impacted by any of the risks disclosed under “Risk Factors—Risks Related to Our Business and Our INVU Platform”, “—Risks Related to Government Regulation and Our Industry” and —Risks Related to Israeli Law and Our Operations in Israel.”
Continue to scale our operations in the United States to accelerate the adoption of our INVU platform. We believe we have assembled a core operating infrastructure to support our future growth. For example, we have a seasoned management team based across Israel and the United States, as well as a comprehensive portfolio of intellectual property and strategic relationships with key suppliers, which we believe position us to rapidly grow our operations. We expect to scale our business in the United States by hiring additional U.S.-based managers as well as sales and marketing, product specialist and end-user support personnel to enhance our ability to acquire customers and retain and grow these relationships. As we grow, we intend to continue to remain asset-light by relying on a network of third-party suppliers and manufacturers to produce our hardware solutions, including our proprietary wireless sensor bands, and to clean and fulfill new orders for our wireless sensor bands.
Build a growing user and partner base through a stepwise approach, from providers to payers, while investing in expanding awareness of our INVU platform. We expect strategic partnerships with care providers to increase traction for our services and allow us to scale more quickly. We believe we have a healthy pipeline of U.S. and international providers and payers of strategic relevance, which we believe we will be able to convert to long-term partners and customers over time. In addition, we intend to spend considerable time and resources seeking to educate expectant mothers and their clinicians about the benefits of our remote monitoring technology. We intend to leverage content creation, advertising, social media and other marketing mechanisms to increase awareness of our solutions among expectant mothers. To increase awareness of our solutions among clinicians, prenatal care providers and other medical professionals, we intend to participate in industry conferences, advertise in medical journals and seek and promote customer testimonials and payer recommendations. We expect that increased awareness among these groups will highlight the benefits of our INVU platform, including ease of use, cost savings, access, and quality of data, which should increase adoption and accelerate our growth.
73
Aggregate the data we capture to enable us to effectively utilize our actual and potential data-related competitive advantages to benefit our user community and population health in general. Collecting data from users on our INVU platform will provide us with significant data that has not been previously captured, either in-office or remotely, about the different stages of pregnancy for both the unborn baby and expectant mother. We expect to expand the data we collect by seeking clearance to extend our INVU monitoring period. Aggregating such data will enable us to effectively utilize the competitive advantages our data collection and analysis capabilities provide to, for example, identify patterns and trends that are associated with certain risks and outcomes from which we should be able to make highly useful and actionable recommendations to expectant mothers and their clinicians.
Continue investing in research and development to enhance the quality and performance of our INVU platform. We have spent considerable time and resources developing our INVU platform and its enhanced system of remote pregnancy monitoring, as well as the intellectual property protecting it. We believe that continued investment in our research and development capabilities will enable us to obtain additional regulatory clearances to support the expansion of our service offerings from our INVU platform. We believe we are one of a few remote providers to be able to capture, compute and visualize this data to clinicians. A comparative study that we have conducted demonstrated that our measurements of MUA are substantially equivalent to those taken with an intrauterine pressure catheter (“IUPC”), a device used during labor to measure the frequency, duration and strength of uterine contractions and which is considered to be the most accurate for MUA measurements. Next, we plan to develop and utilize the measurements within our capabilities, such as mECG, fECG, fPCG, mPCG, fetal activity, fetal position, maternal respiration and amniotic fluid volume, to power and fuel our predictive model. We may also expand our offerings by seeking clearance to provide some of these measurements to expectant mothers and clinicians. We expect that continued investment in research and development will allow us to improve our product offerings and enable our products to become the standard of care for remote pregnancy monitoring and pregnancy care management. We believe that maintaining and growing our intellectual property portfolio will protect and expand our competitive position. See “—Our Competitive Advantages” and “—Research and Development.”
We intend to expand our reach globally. Our ambition is to improve pregnancy care globally. We filed for a CE mark in Europe in March 2023, which if approved, should allow us to offer NSTs using our FHR, MHR and MUA capabilities in certain circumstances. Our business development work in this region indicates substantial demand for our solutions. We are in discussions already in the Netherlands, Germany and Israel with various enterprise-level healthcare systems as well as payer networks. In Germany we have established a partnership with Charite University to bring the previously established benefits of remote pregnancy monitoring to Europe for the first time, while also setting the stage for the use of predictive analytics to improve health outcomes in the future. Finally, we are also involved with and pursuing a relationship with one of the four leading health maintenance organizations (“HMOs”) in Israel that is an innovator in remote marketing, and we also have a relationship with a top ranking Israeli medical center that is a global leader in medical innovation. If we obtain clearances and approvals in these and other jurisdictions, we believe our expanded reach would allow us to become a leader in pregnancy solutions from the first days of pregnancy onward. If we are able to scale globally, we expect to maintain our fundamental approach to commercialization to focus on building strong relationships with local care networks and payers as our anchor partners.
Our Challenges
We face company and market challenges to meet our objectives, including the following:
| ● | Corporate growth: we are a company headquartered in Israel with the development of our INVU technology far from where it will be commercialized. Product-market match is a difficult endeavor anywhere, but particularly when development is done far from where it will be applied. Building a commercial team and operations in a new market far from headquarters is a challenge, but one that is growing with a US-based CEO, CFO, Vice President, product specialists, and customer support. The majority of the new hires planned for 2024 will be in the U.S. market to support growth targets. See “—Research and Development”, “—Manufacturing and Supply” and “—Sales and Marketing”. |
| ● | Product development: moving from prototype to mass manufacturing is difficult, as will working to continue reducing costs over time. In addition, we may continue to face challenges to ensure that upstream innovation will be able to be commercialized within regulatory and reimbursement frameworks. |
| ● | Regulatory approvals: the pre-natal market is relatively new to in-home monitoring. The risks of the population – the unborn baby and expectant mother – presents additional challenges and sparks extra review by the FDA. Safety record, a lack of adverse events and reports, among others, can help allay issues with respect to regulatory approvals in this space. See “—Government Regulation” and “Risk Factors—Risks Related to Government Regulation and Our Industry”. |
| ● | Competition: there are several competitive devices/solutions that are aiming for commercialization. We believe this is a validation of the commercial opportunity. The vast majority of these new competitors seem to be doing away with traditional Doppler/TOCO technology and attempting to monitor with direct physiological signals, such as biopotential, which is further validation of Nuvo’s direction. However, this is something to be mindful of because competitors are fast approaching with the ability to collect similar signals, and we will have to continue to innovate and execute effectively to achieve our objectives. See “—Competition.” |
74
Recent Developments
Bridge Financing
Since November 2023 Nuvo has been engaged in a bridge financing (the “Bridge Financing”), which involves the issuance of secured convertible bridge notes (individually, a “Bridge Financing Note”; collectively, the “Bridge Financing Notes”) to investors (“Bridge Financing Holders”).
The Bridge Financing Notes carry a 15% annual interest rate and upon conversion on the applicable Maturity Date (as defined in the Bridge Financing Notes), (i) Nuvo will pay the Holders all accrued interest on the Bridge Financing Notes up to the date of payment or conversion, and (ii) the Holders in their sole discretion, may choose to either (a) receive the principal amount of the Bridge Financing Note in cash; or (b) convert the principal amount of the investment into Nuvo Shares at a price per share of $7.0265 (or, post-Closing, the resulting number of Holdco Ordinary Shares after applying the equity exchange ratio of 96.139%).
As of the date hereof, approximately $8.5245 million in principal amount of Bridge Financing Notes has been received by Nuvo, and the offering of the Bridge Financing Notes remains ongoing.
From March 24, 2024 through April 8, 2024, Nuvo entered into amendments to all of the existing Bridge Financing Notes representing $6.5732 million principal amount of the Bridge Financing Notes, to extend the maturity dates thereof (the “Bridge Financing Notes Amendments”). All new Bridge Financing Notes since April 8, 2024 include the amended maturity definition. Prior to the Bridge Financing Notes Amendments, the Bridge Financing Notes were scheduled to mature on the earlier of (i) twelve months from the issuance date thereof, (ii) the closing of the Business Combination, (iii) the closing of an initial public offering, or (iv) the closing of a bona fide financing by Nuvo for the principal purpose of raising capital, through the sale of Nuvo securities in whatever form or type (whether debt or equity) that raises in excess of $10,000,000 in gross proceeds. Pursuant to the Bridge Financing Notes Amendments, the maturity date of the amended Bridge Financing Notes was revised to be the earlier of (i) twelve months from the issuance date thereof, (ii) six (6) months following the closing of the Business Combination, (iii) six (6) months following the closing of an initial public offering, or (iv) the closing of a bona fide financing by Nuvo for the principal purpose of raising capital, through the sale of Nuvo securities in whatever form or type (whether debt or equity) that raises in excess of $25,000,000 in gross proceeds.
Each Bridge Financing Note is secured by all of Nuvo’s intellectual property, and Nuvo has filed collateral assignments/financing statements with the United States Patent & Trademark Office and is in the process of filing collateral assignments/financing statements with Nuvo’s Registrar in Israel. Gaingels 10x Capital Diversity Fund I, LP, a Bridge Financing Holder and an affiliate of a member of the Sponsor serves as collateral agent with respect to the collateral securing the Bridge Financing Notes. Upon the occurrence of any event of default described therein, the outstanding balance under the Bridge Financing Notes shall become immediately due and payable upon election of the Bridge Financing Holder and following a written demand notice sent to Nuvo.
In consideration for the services to be rendered under certain advisory services agreements between the Bridge Financing Holders and Nuvo, Nuvo issued a warrant to each Bridge Financing Holder, whereby the Bridge Financing Holder is given the right to purchase such number of Nuvo Shares (or, post-Closing, Holdco Ordinary Shares after applying the equity exchange ratio of 96.139%) equal to (2x) the principal amount of the Holder’s Bridge Financing Note divided by the same price per share noted above (i.e., $7.0265), at an exercise price of NIS 0.01.
This summary is qualified in its entirety by reference to the full text of each of the form of Bridge Financing Convertible Note, the form of Bridge Financing Warrant and the form of Bridge Financing Notes Amendment, which are filed as exhibits 4.10, 4.11 and 4.12, respectively.
75
Expanded Commercial Partnership
Philips
We and Philips Electronics Nederland B.V., or Philips, entered into a master purchase agreement (“Philips MPA”), in August 2023, the scope of which consists of Philips or one of its affiliates procuring products and services from Nuvo. Products include Nuvo’s proprietary INVU remote patient maternity monitoring system, or the INVU System, and services include professional services rendered by Nuvo in connection with Nuvo’s delivery of the INVU System. The aggregate amount paid under the Philips MPA to date is $0. The designated territory for this commercial arrangement is the United States (the “Territory”).
Other important terms under the Philips MPA include:
| ● | Nuvo has granted Philips limited exclusivity to market the INVU System to certain market segments. |
| ● | The parties have agreed to create a steering committee made up of three senior representatives from each organization. The committee will be tasked with various governance-related responsibilities, such as overseeing the progress of the parties; generally planning and scheduling; considering any proposed changes or modifications to the Philips MPA; conflict resolution; and such other matters as may be agreed between the parties from time to time. |
| ● | The pricing model involves essentially an all-in price (i.e., the ‘transfer price’) per pregnancy which includes delivery of the INVU sensor band, access/use of the INVU solution, and the support provided by Nuvo. Professional services, if any, that Nuvo may be requested to carry out will be charged separately. |
| ● | Each party will maintain ownership of its respective pre-existing intellectual property, as well as any modifications made to such intellectual property during the course of the agreement. The Philips MPA contains certain provisions protecting Nuvo’s intellectual property and confidential information in a commercially reasonable manner. |
| ● | The Philips MPA has a five-year initial term, unless either party terminates it after the first three years upon 180 days’ notice to the other party, upon a commercially reasonable basis. |
This Philips MPA was preceded by a Master Pilot Agreement that was signed in 2021. Since then, Nuvo and Philips have activated different workgroups that meet regularly and cover all aspects of the partnership, including integration, marketing, regulatory, order management, and contracting/supplier qualification. All paperwork behind the joint offering integration is complete in the form of an amendment to the Master Pilot Agreement that was signed in January 2023. Nuvo believes that the parties are currently on track to activate their vision of distribution of the joint offering to hospital networks in the United States within the next six months.
76
Our Market Opportunity
According to the National Institutes of Health/American Medical Informatics Association data13, the United States is spending over $110 billion annually on prenatal and neonatal care, which is growing at approximately 3% per year for public and private insurance. In the United States, there are 3.66 million annual pregnancies14 of which 38% are treated as low risk and 62% are treated as high risk, according to an AJOG study.15 According to a 2020 JAMA Network article16 covering changes in U.S. health care spending by payer and health condition over time, general pregnancy costs represent approximately $53 billion of spending and high-risk pregnancies represent approximately $58 billion of additional spending. Furthermore, according to a 2020 article by the American Journal of Managed Care17 covering the costs of giving birth in the United States, the average national cost of childbirth admission for an individual with employer-sponsored insurance is approximately $14,000. We believe that this large, dynamic market is ripe for disruption for several reasons in addition to the shortage of clinicians. First, the rise in adverse outcomes for infants and mothers, including increased mortality and an increase in HRPs, requires better solutions. Second, current fetal monitoring technology is outdated, as it is primarily based upon CTG, used to monitor fetal heart rate and uterine contractions during pregnancy and labor, which was designed for intrapartum monitoring in clinics by experienced professionals. Existing devices for fetal monitoring in clinical use today, such as the in-clinic Philips Avalon FM-30 and the GE Corometrics 250 Series, or the home use device Sense4Baby, rely on doppler for recording the fetal heart rate, and TOCO for recording uterine contractions.
According to an article in the Journal of Obstetric, Gynecologic & Neonatal Nursing18, a well-documented risk of Doppler technology is that it may acquire a fetal heart rate mistakenly from the mother’s heart-beat, potentially leading to false reassurance. Such false reassurance may lead in turn to results such as stillbirth, according to an article published in the British Medical Journal19 discussing the dangers of listening to the fetal heart at home. In addition, if the signals are not differentiated correctly, maternal decelerations can be mistaken for fetal decelerations, leading to unnecessary intervention. The medical community has acknowledged this problem and we think active measures should be taken to avoid it. In the INVU device, the measurement of biopotential signals coming directly from the fetal and maternal hearts, and the combination of biopotential and acoustic signals is designed to differentiate the fetal from the maternal heart rate. In addition to the clinical data presented below under “Clinical Studies”, this capability has been demonstrated internally via bench testing where several scenarios of simultaneous MHR and FHR simulated signals (including overlapping MHR and FHR signals) were provided as input to the INVU system and the INVU system successfully distinguished between the two signals without error. Furthermore, according to a comparative study found in the Gynecologic and Obstetric Investigation Journal20, FHR and MHR recordings using an ECG-based system have been reported to significantly reduce the rate of confusion between FHR and MHR compared to the same data recorded via CTG.
According to a study published in the AJOG21, comparing three methods of monitoring uterine activity during labor, TOCO, utilized in CTG to record maternal contractions, has been shown to have a high miss rate, potentially up to 35%, when compared to IUPC. In contrast, the INVU system has been shown to have a positive agreement of 85% (meaning a miss rate of about 15%) when compared to IUPC (see results below under “Clinical Studies”). Within the same study, INVU and TOCO were recorded simultaneously and INVU significantly outperformed TOCO.
As a result of the above, we believe that the INVU system is more reliable than the current monitoring solutions generally provided in healthcare facilities. Taken together with the ability to accurately record FHR, MHR and MUA remotely via a self-administered device, we believe that the INVU system is better equipped to remotely monitor many pregnancy-related problems during the antepartum period.
Third, the substantial costs of pregnancy care continue to increase without any meaningful corresponding improvement in results and, in some cases, results in a failure to receive care. Fourth, the United States is facing a growing and severe shortage of clinicians during a time when better care is needed. Healthcare has been trending to telehealth infrastructure and remote monitoring solutions for care delivery and practice revenue generation. The COVID-19 pandemic also greatly accelerated the move to telehealth.
| 13 | American Medical Informatics Association, “Overcoming the Maternal Care Crisis: How Can Lessons Learnt in Global Health Informatics Address US Maternal Health Outcomes?”, AMIA Annual Symposium Proceedings Archive, Article (Published online April 16, 2018). |
| 14 | US Centers for Disease Control and Prevention, National Vital Statistics Report, “Births: Final Data for 2021”, Volume 72, Number 1 (January 31, 2023). |
| 15 | American Journal of Obstetrics and Gynecology, “Unexpected complications of low-risk pregnancies in the United States”, Volume 212, Issue 6, Article (June 2015). |
| 16 | JAMA Network, “US Health Care Spending by Payer and Health Condition, 1996-2016”, Article (March 3, 2020). |
| 17 | American Journal of Managed Care, “How Much Does It Cost to Give Birth in the United States? It Depends on the State”, Article (May 15, 2020). |
| 18 | Journal of Obstetric, Gynecologic & Neonatal Nursing, “Maternal or Fetal Heart Rate? Avoid Intrapartum Misidentification”, Volume 33, Issue 1, Article (January 2004). |
| 19 | British Medical Journal, “The dangers of listening to the fetal heart at home”, Volume 339, Article (November 14, 2009). |
| 20 | Gynecologic and Obstetric Investigation Journal, “Intrapartum heart rate ambiguity: a comparison of cardiotocogram and abdominal fetal electrocardiogram with maternal electrocardiogram”, Volume 75, Issue 2, Article (January 2013). |
| 21 | American Journal of Obstetrics & Gynecology, “Monitoring uterine activity during labor: a comparison of three methods”, Volume 208, Issue 1, Article (January 2013). |
77
We believe Nuvo’s approved product can immediately address the following market opportunities:
| ● | Remote NSTs: According to the ACOG, current clinical practice calls for performing a NST once or twice weekly after the 32nd week of pregnancy22. As noted below (see Figure 1), 38% of all pregnancies in the United States are treated as low-risk and 62% are treated as high-risk, according to a study in the AJOG analyzing approximately ten million pregnancies. Given that 62% of the approximately 3.66 million annual pregnancies in the United States23 are treated as high-risk, and given that current clinical guidelines call for once or twice weekly NSTs from gestational age 32 to term, on average, at least approximately 18 million NSTs are conducted annually in the United States alone, although this number could reach as high as approximately 36 million considering the common clinical practice of twice-weekly NSTs. An expectant mother with a high-risk pregnancy can perform any NST prescribed by her clinician at home, in lieu of an office visit. The total yearly NST market in the United States has the potential to be in the range of approximately $1.5 billion to $3.2 billion. Nuvo’s current capabilities are poised to capture a significant share of this market, offering cost-effective, remote monitoring solutions. We believe by taking our current list price or average selling price of our INVU platform and multiplying it by the 3.66 million annual pregnancies in the United States, we can estimate our total addressable market. We are targeting capturing approximately 10% of this market in the United States over the next several years, the entirety of which we believe is a serviceable market for Nuvo on its existing FDA-cleared product capabilities alone. |
| ● | Spontaneous OB-Emergency Department (OB-ED) Visit Reduction: According to a study contained in the AJOG in 201724, approximately 36% of all pregnant women visit the OB-ED at least once during their pregnancy. Nuvo’s current solution can potentially decrease OB-ED visits by offering timely and accurate home-based assessments, which is currently valued at approximately $2.4 billion annually. For example, Nacogdoches Women’s Center, a private practice based out of Texas and customer of Nuvo, has cited that their patients are avoiding spontaneous OB-ED visits because they are triaged remotely (i.e. used INVU for one-time, on-demand remote monitoring towards effective assurance to assuage concerns about decreased fetal movement or other concerning factors that would have otherwise required hospital presentation), further validating Nuvo’s belief that it can currently address this market opportunity and in the future, more fully address this market opportunity as Nuvo acquires more data and monitoring of that data to support this claim definitively. In addition, future products, subject to FDA approvals, may further Nuvo’s claims on this topic. |
| ● | Maternal Hospital Admission/Reducing Length of Stay: According to a study in Denmark25 reporting on 400 singleton pregnancies, home-monitoring, including remote self-monitoring of fetal and maternal well-being in intermediate- and high-risk pregnancies appeared to be a safe, feasible alternative to inpatient or frequent outpatient care, improving expectant mother well-being and cost-effectiveness. The Denmark study used remote monitoring solutions to validate the solution, albeit solutions that do not have on-label indication for home use (specifically, the Monica Novii patch). Home hospitalization was conducted on otherwise hospitalized inpatients, such as those with preeclampsia and PPROM (preterm premature ruptured membranes). Daily monitoring was conducted, for days or weeks. Findings included no adverse health outcomes as compared to traditional hospitalization, with improved cost outcomes due to reduced hospitalization days. Reducing hospital admissions and length of hospital stay is a benefit that can currently be provided with Nuvo’s FDA-cleared solution. Moreover, our future AI and biomarker enhancements may enable earlier detection of potential complications, optimizing the need for hospital admissions and potentially reducing the length of stay even further. These abilities give Nuvo reason to believe it can capture a portion of the current $38 billion spent in this arena (according to data from a 2019 study from the Journal of Pediatrics26 covering birth hospitalization costs and days of care for mothers and neonates in California and taking into account inflation rates). |
| ● | Reducing Unnecessary C-Section Rate: With advanced predictive capabilities, Nuvo aims to lower the number of unnecessary C-sections through better monitoring and insights. This shift would not only represent improved health outcomes but also aligns with reducing a portion of the nearly 1.2 million C-sections performed annually in the United States according to the CDC27. In addition, members of Nuvo’s Medical Advisory Board, comprised of seven members consisting of professors, physicians and experts with years of experience in women’s health, obstetrics, gynecology and fetal medicine, believe Nuvo’s currently available INVU solution may already help reduce unnecessary C-sections, by theorizing that remotely monitoring expectant mothers who otherwise would be coming in the hospital for induction and thus, will help reduce labor fatigue. This use case has not yet been implemented in practice yet as this is a benefit that may be provided with our FDA-cleared solution, but will require more data and monitoring of that data to support this claim definitively. In addition, future products, subject to FDA approvals, may further Nuvo’s claims on this topic. |
| 22 | The American College of Obstetricians and Gynecologists, “Indications for Outpatient Antenatal Fetal Surveillance”, ACOG Committee Opinion, Committee on Obstetric Practice Society for Maternal-Fetal Medicine, Volume 137, Number 828 (June 2021). |
| 23 | US Centers for Disease Control and Prevention, National Vital Statistics Report, “Births: Final Data for 2021”, Volume 72, Number 1 (January 31, 2023). |
| 24 | American Journal of Obstetrics & Gynecology, “Non-Urgent and Urgent Emergency Department Use During Pregnancy: An Observational Study”, Volume 216, Issue 2, Article (February 2017). |
| 25 | Acta Obstetricia et Gynecologica Scandinavica, “Home management by remote self-monitoring in intermediate- and high-risk pregnancies: A retrospective study of 400 consecutive women”, Volume 101, Issue 1, Article (January 2022). |
| 26 | Journal of Pediatrics, “Birth Hospitalization Costs and Days of Care for Mothers and Neonates in California 2009-2011”, Volume 204 (January 2019). |
| 27 | US Centers for Disease Control and Prevention, “Births – Method of Delivery”, Article (Last reviewed June 8, 2023). |
78
| ● | Upstream Innovation Benefits: Nuvo intends to utilize AI for predicting and reducing preterm births ($28 billion annual cost according to JAMA28), maternal mood and anxiety disorders (which can cause an annual economic impact of approximately $14 billion according to a study discussed in a 2019 Mathematica article29, with $2.9 billion directly related to maternal health expenditures), and neonatal intensive care unit stays for newborns ($24 billion annual cost according to a 2018 article in the Journal of Pediatrics30). These areas present a market opportunity that aligns with our mission to improve healthcare outcomes and equity. Nuvo’s future innovations, subject to FDA approvals, intend to address commercial market opportunities in these segments. |
According to the World Health Organization (“WHO”)31, globally, approximately 295,000 women died from preventable causes related to pregnancy in 2017, or approximately 810 women every day. According to a study published in 201732, approximately 36% of the pregnant women who participated in the study had at least one visit to the emergency department that was non-urgent. While the WHO33 has recommended that C-sections should not exceed 10% to 15% of all deliveries in any country, the CDC34 reported approximately 32% of all deliveries in the United States were by C-section in 2018. According to the CDC35, pre-term birth affected about one of every ten infants born in the United States in 2022.
Figure 1:

| 1. | CDC 2021, Vital Statistics Rapid Release |
| 2. | AJOG June 2015, Unexpected complications of low-risk pregnancies in the United States |
| 3. | JAMA 2020, US Health Care Spending by Payer and Health Condition, 1996-2016 |
| 4. | AAP 2017, Guidelines for Perinatal Care, 8th Edition |
| 5. | CDC 2021, Births – Method of Delivery |
| 6. | CDC 2021, Gestational Diabetes |
| 7. | CDC 2021, High Blood Pressure During Pregnancy |
| 8. | Reproductive Toxicology 2021, Intrauterine growth restriction: Clinical consequences on health and disease at adulthood |
| 9. | Medscape 2016: Premature Rupture of Membranes |
| 28 | JAMA Network, “US Health Care Spending by Payer and Health Condition, 1996-2016”, Article (March 3, 2020). |
| 29 | Mathematica, “New Study Uncovers the Heavy Financial Toll of Untreated Maternal Mental Health Conditions”, Article (April 29, 2019). |
| 30 | Journal of Pediatrics, “Variation in Use by NICU Types in the United States”, Volume 142, Issue 5, Article (November 1, 2018). |
| 31 | World Health Organization, Department of Reproductive Health and Research, Evidence brief, Article (2019). |
| 32 | American Journal of Obstetrics & Gynecology, “Non-Urgent and Urgent Emergency Department Use During Pregnancy: An Observational Study”, Volume 216, Issue 2, Article (February 2017). |
| 33 | World Health Organization, “WHO statement on caesarean section rates”, Article (April 14, 2015). |
| 34 | US Centers for Disease Control and Prevention, “Births – Method of Delivery”, Article (Last reviewed June 8, 2023). |
| 35 | US Centers for Disease Control and Prevention, Reproductive Health, “Preterm Birth”, Article (Last reviewed October 24, 2023). |
79
Adverse Maternal and Infant Outcomes
The ability to protect the health of mothers and babies in childbirth is generally considered to be one of the fundamental indicators of a society’s development. According to the World Population Review36, the United States ranks 50th in the world for infant mortality, with 5.4 deaths per 1,000 live births, making it one of the most dangerous developed countries to give birth in, behind many emerging countries. These troubling trends are amplified by a continuous increase in the number of HRPs and dangerous co-morbidities such as diabetes and hypertension, which disproportionately affect women in rural communities and of lower socioeconomic status. The March of Dimes reported in 202237 that nearly 36% of U.S. counties were “maternity care deserts” or counties where there were no hospitals providing obstetric care, no birth centers, no OB/GYN and no certified nurse midwives. While the overall rate of infant mortality has declined, the United States continues to lag behind other developed countries. According to the most recent data from the Organization for Economic Co-operation and Development (“OECD”)38, babies in the United States are 76% less likely to reach their first birthday than babies born in other wealthy member countries.
While most women in the United States give birth to healthy babies and without serious complications, pregnancy and childbirth come with a variety of health risks for both mother and baby. A growing number of challenges facing care of the expectant mother and unborn baby in the United States are helping to drive these troubling trends. An increasing incidence of chronic conditions, such as obesity, diabetes and hypertension, among expectant mothers, is driving up rates of HRP. According to the CDC39, one in every 12 to 17 pregnancies among women ages 20-44 is hypertensive. According to Medscape40 and the Journal of the American Medical Association41, costs related to hypertension are approximately $15,000 on average and result in annual spending of $5.5 billion.
Increased monitoring of the pregnancy’s progression, which enables quicker and earlier intervention, is critical to protecting the health of mother and baby. However, limitations in the portability, effectiveness and administration of current monitoring technology means that even with additional prenatal visits, insight into HRP remains fragmented and makes charting a path towards better outcomes challenging. HRPs pose significant risk to mother and baby and also come at a high cost for both parents and the healthcare system.
Outdated Practices and Tools
In many areas of medicine, advancements in technology have made healthcare solutions commonplace that only a few decades ago seemed impossible. Digitization of healthcare and the shift of a portion of care delivery to the home, in general, is progressing at a rapid pace. Unfortunately, while a significant number of practice areas, such as oncology, cardiovascular medicine and chronic conditions, have seen substantial technological innovation, innovation with respect to the health of the expectant mother and the unborn baby has been slower. In fact, the technology, which has changed little in more than three decades, is outdated and inefficient and has not developed fundamentally new, scalable and remote approaches to pregnancy monitoring and care, resulting in an overburdened healthcare system in which parents and clinicians have minimal visibility to what is actually happening in the pregnancy.
CTG, the monitoring technology underpinning, in most cases, today’s standard for pregnancy management, was designed and built for intrapartum pregnancy monitoring by a medical professional in a hospital setting. CTG lacks practical portability and requires active in-office administration by a medical professional as it requires expertise to apply the device and maintain it in a proper position. CTG also utilizes Doppler and TOCO technologies that only allow for active indirect and passive indirect, respectively, average measurements of heart rate and uterine contraction activity. Accordingly, this approach is generally limited to the information about the health of a pregnancy that can be collected during monthly or less often in-office visits, or, in the case of HRPs, for example, far more often.
| 36 | World Population Review, “Infant Mortality Rate by Country”, Article (Updated 2024). |
| 37 | March of Dimes, “Nowhere to Go: Maternity Care Deserts Across the U.S.”, Maternity Care Deserts 2022 Report. |
| 38 | Health Affairs, “Child Mortality In The US and 19 OECD Comparator Nations: A 50-Year Time-Trend Analysis”, Volume 37, No. 1, Article (January 2018). |
| 39 | US Centers for Disease Control and Prevention, “High Blood Pressure During Pregnancy”, Article (Last reviewed June 19, 2023). |
| 40 | Medscape, “Hypertension and Pregnancy”, Article (June 22, 2022). |
| 41 | JAMA Network, “US Health Care Spending by Payer and Health Condition, 1996-2016”, Article (March 3, 2020). |
80
Despite the large number of global births, existing remote pregnancy monitoring technology has certain weaknesses and has not been able to solve many of the problems facing pregnancy care today, which amplifies the need for technology such as ours, which consists of an innovative sensor form factor and computations resulting in higher quality signals. Existing systems are generally less accurate and have difficulty separating FHR from MHR. Neither are they able to measure heart rate beat-by-beat or heart rate variability. As indicated, we obtain both a biopotential signal and an acoustic signal, which allows us to measure heart rate beat-by-beat and heart rate variability, and to capture other layers of data. INVU is also able to sense movement of the expectant mother or unborn baby, providing significant advantages as to the data we are able to collect, although it does not currently provide movement data to the clinician or expectant mother. Few existing remote pregnancy monitoring technology solutions measure MUA, meaning that, currently, this measurement must almost always be done at the clinician’s office, a hospital or similar facility. INVU also takes all measurements passively, or from the body, as opposed to going through the body, as is the case with Doppler ultrasound which is an active technology that sends signals into the womb. Current remote technology that requires placement on the body either cannot be self-administered or self-administration is difficult due to the movement of the transducers, and as the amount of pressure utilized during this procedure varies, results vary. Notwithstanding, even the existing remote pregnancy monitoring has been shown in limited analysis to increase monitoring compliance, improve quality of care and healthcare outcomes and reduce payer costs. Accordingly, we believe that our advanced technology will be even more successful in this regard.
Utilizing outdated technology, parents and clinicians receive a fragmented and episodic view of the pregnancy that makes it difficult to confidently chart the course of a pregnancy and identify potential problems that could impact the health of the expectant mother and unborn baby. Additionally, the pregnancy management process could consume significantly more travel and work time for parents and cost for payers and uninsured expectant mothers, all of which could have an adverse effect on compliance. Despite the serious implications of complications during pregnancy, to date, we believe that a holistic remote platform for integrated, connected and continuous care has not been available until INVU.
Substantial Costs of Pregnancy Care
Another factor affecting access is that for all pregnancies, the cost to give birth in America has been increasing without any corresponding improvement in results. Many expectant mothers are forced to pick and choose care based on their ability to pay or insurance, rather than seek the best available medical care. According to The Commonwealth Fund42, more than one-third of women in the United States report skipping needed medical care because of costs. According to the U.S. Department of Health and Human Services Office on Women’s Health43, babies of mothers who do not get prenatal care are three times more likely to have a low birth weight and five times more likely to die than those born to mothers who do get care. In addition, approximately 36% of all pregnant patients who participated in a study44 presented at the OB emergency department (OB-ED) at least once during their pregnancy. Nuvo’s solution is being used to provide remote triage and monitoring to expectant mothers instead of going to the OB-ED, saving cost, time and effort for expectant mothers, providers, and payers. These cost issues which affect access can also be traced back, in part, to the outdated technology and standard of care. The increase in HRPs has also increased the costs of pregnancies. Thus, the need for more affordable and accessible care is significant.
| 42 | The Commonwealth Fund, “What Is the Status of Women’s Health and Health Care in the U.S. Compared to Ten Other Countries?”, Article (December 19, 2018). |
| 43 | U.S. Department of Health and Human Services, Office on Women’s Health, “Prenatal care”, Article (Last updated February 22, 2021). |
| 44 | American Journal of Obstetrics & Gynecology, “Non-Urgent and Urgent Emergency Department Use During Pregnancy: An Observational Study”, Volume 216, Issue 2, Article (February 2017). |
81
Severe Shortage of Obstetrician Access
Despite rising birth rates and cases of serious HRPs, as previously indicated, access to obstetrics services continues to decline, especially in rural communities. This is due in large part to a growing shortage of obstetricians. The ACOG45 puts the current shortage at approximately 9,000 obstetricians countrywide and expects this number to grow to 22,000 by 2050, in part because the average age of obstetricians is relatively high, and a number of obstetricians are nearing the end of their career. The March of Dimes reported in 202246 that nearly 36% of U.S. counties were “maternity care deserts” or counties where there were no hospitals providing obstetric care, no birth centers, no OB/GYN and no certified nurse midwives. Not only is this burden being felt by the dwindling numbers of obstetricians, but a growing number of families are being put at a distance from care that is inconvenient, and at least in some cases, dangerous. Women who live in these counties may also have limited access to appropriate preventive, prenatal and postpartum care. Distance can make the recommended number of office visits difficult to comply with. The shortage of obstetricians can result in fragmented, impersonal care that does not reflect what research has shown produces the best health outcomes for mothers and babies. Even in counties with obstetricians, expectant mothers are reporting seeing a reduction in total time spent in prenatal visits.
One of the largest contributing factors to this decline is the overburdening of obstetricians, many of whom struggle to manage mounting numbers of expectant mothers to care for with unpredictable hours and high liability potential. Rather than offer a solution, standard pregnancy care technology often exacerbates these issues. Current clinical guidelines according to the American Academy of Pediatrics47, recommend about 11-15 prenatal visits over the course of a healthy pregnancy. Most of these visits simply confirm that the pregnancy is progressing as expected, but limitations in monitoring often mean that most of these visits must be done in-office with expensive technology regardless of the status of the pregnancy. Current ACOG clinical guidelines48 suggest once or twice weekly NSTs for HRPs in the last trimester. With these limitations and episodic care, almost 75% of expectant mothers who participated in a study49 made an unscheduled obstetrics visit, with 38% making two or more unscheduled visits. This is reflective of the unnecessary utilization of resources for clinicians, expectant mothers and health systems. The reduction in the professional labor force available to treat a steadily growing population requires more efficient pregnancy monitoring technology to ensure continuous care.
| 45 | American Journal of Managed Care, “Physician Shortage Likely to Impact OB/GYN Workforce in Coming Years”, Article (September 21, 2019). |
| 46 | March of Dimes, “Nowhere to Go: Maternity Care Deserts Across the U.S.”, Maternity Care Deserts 2022 Report. |
| 47 | American Academy of Pediatrics, Guidelines for Perinatal Care, Eighth Edition, Book (2017). |
| 48 | The American College of Obstetricians and Gynecologists, “Indications for Outpatient Antenatal Fetal Surveillance”, ACOG Committee Opinion, Committee on Obstetric Practice Society for Maternal-Fetal Medicine, Volume 137, Number 828 (June 2021). |
| 49 | American Journal of Obstetrics and Gynecology, “Prenatal health care beyond the obstetrics service: Utilization and predictors of unscheduled care”, Volume 198, Issue 1, Article (January 2008). |
82
Technology
Overview
Our INVU platform begins with a self-administered, wireless sensor band, containing passive biopotential acoustic and motion sensors. Our wireless sensor band’s multiple sensors acquire granular signals that are designed to overcome variability in body physique or build or changes in fetal position. The sensors acquire underlying fetal and maternal ECG and PCG data, along with motion data, which allow for beat-by-beat precision of heart rate calculations. This data is transmitted to our cloud computing back-end application which utilizes two modules of algorithms, one for FHR and MHR and another for MUA, to perform the set of signal processing and analysis tasks needed to extract the clinically meaningful physiological measurements, such as FHR, MHR and MUA, from the raw data. Our multi-modality technology enables robust discrimination between FHR and MHR, separating the data into two corresponding channels for computation and visualization, as well as reliable measurements of MUA when compared to the existing standard of care. The algorithm also displays the data we are cleared by the FDA to provide for visualization through one of two mobile applications that provide tailored information for each of the clinician and expectant mother. The first, a software application for the expectant mother, runs on her mobile device, which displays the results, controls our wireless sensor band and is utilized as a bridge to send the raw data collected to the cloud-based servers for analysis. The other, a software application for the clinician, runs as a web-based application, our INVU Pro application, on any computer with a standard internet browser.
The photograph and diagram below show our wireless sensor band as worn by an expectant mother and a diagram of our wireless sensor band. As can be seen from the diagram, the band consists of an upper and lower band placed at the top and bottom of a woman’s abdomen and connected in the back. Our wireless sensor band contains biopotential, acoustic and motion sensors, some of which are fixed and some of which are removable for ease of repair and replacement.
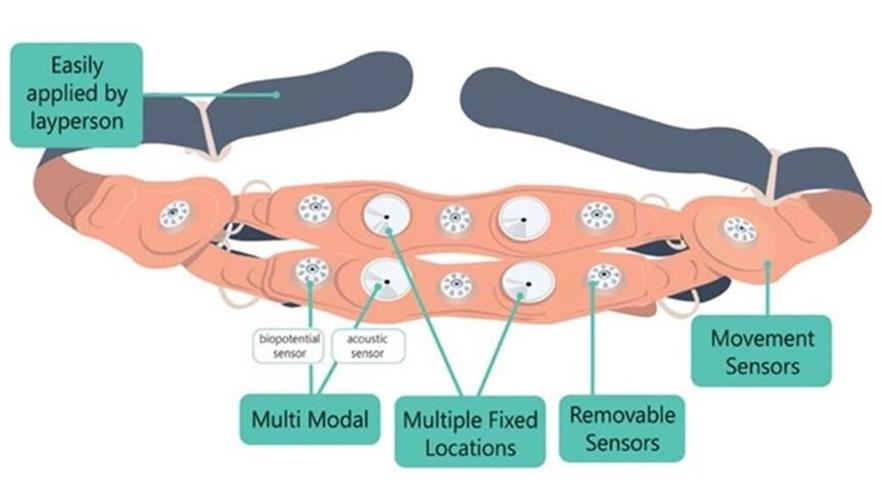
83
Wireless Sensor Band and Sensors
We designed our wireless sensor band to be easy to administer by a lay person, such as an expectant mother or her significant other. Our wireless sensor band consists of eight biopotential sensors, four acoustic sensors and two motion sensors overlaid on a lightweight but durable band. The biopotential sensors measure small potential or voltage changes on the skin that arise from physiological signals, including the cardiac biopotential signals generated during each heartbeat. The acoustic sensors are highly sensitive microphones that convert sound waves into an analog biopotential signal. These sensors passively detect the ECG and PCG signals unlike most other existing pregnancy monitoring devices, which actively send signals into the womb and which often require constant readjustments by trained professionals, limiting the ability of those devices to monitor continually within protocol. Motion sensors monitor maternal or device motion which could affect the physiological measurements being taken and helps us to validate our captured data or determine if such data was interrupted by an abrupt movement. We have also developed a real-time module that we expect would notify the expectant mother to stop moving if the movement is interrupting signal acquisition and we are testing using the motion sensor to further validate our MUA algorithm. Raw data from each sensor are converted from analog-to-digital, and sent by Bluetooth to the expectant mother’s mobile device, which transmits the signal securely to the cloud for processing.
MHR and FHR Signal Detection and Processing
After the data are acquired, they are digitized and sent wirelessly for analysis on cloud-based servers by an algorithm we developed. The goal of the algorithm is to fuse the independent information gathered from the acoustic and biopotential sensors to obtain FHR and MHR. The algorithm validates the data, preprocesses the data to remove noise, detects heartbeat independent from biopotential signals and acoustic signals and fuses detected heartbeats from biopotential and acoustic signals to calculate FHR and MHR. Raw data is examined to determine whether it contains valid data. Biopotential data are treated as containing a valid signal if the mECG can be detected because the mECG has a large enough amplitude to appear with adequate quality in all biopotential channels. Acoustic channels that are suspected of containing only noise are considered invalid and discarded.
Acoustic signals and biopotential signals are then independently filtered to capture the relevant physiological signals and to reduce unwanted signals or noise. An additional filter eliminates low-frequency noise in signals with high levels of noise. Acoustic signals are preprocessed with multiple sensor bandpass filters that accept signals within a specific range of 10-95 Hz and rejects signals outside that range.
mECG is determined by detecting maternal Q, R and S waves, or the QRS complex, which are biopotential signals that spread throughout the ventricles in rapid succession when the heart changes from a resting polarized state, or more negative electrical charge, to a depolarized state, or more positive electrical charge, represent maternal heartbeats. These biopotential signals are cross-correlated between multiple ECG channels and for each detected and adjacent heartbeat to enable extraction of the mECG signals from the detected biopotential signals. Once the mECG is extracted from the signal, it is then subtracted from each channel of biopotential data, leaving the fECG data and noise not earlier eliminated. The remaining data are processed to determine fECG by filtering the signal within a pre-determined range of frequencies and then enhancing it to process the resulting signals for fetal QRS detection through peak detection and cross-correlation.
Each channel is examined through the algorithm to determine whether PCG signals represent “true” heart sounds, for example, the heartbeat when (i) the heart muscle contracts after refilling with and pumping blood from the heart chambers into the arteries and (ii) the aortic valve and pulmonary valve close. This is achieved by calculating an outline of the signal extremes, discarding peaks that are not prominent, and grouping peaks into two groups according to shape and size using a clustering algorithm, or an algorithm that estimates density and tends to group the data points belonging to a single distribution together. An initial estimate of the beat-to-beat interval of each PCG group is calculated. Missing beats are identified and added as appropriate. In parallel, the peaks of the PCG signal are auto-correlated. The algorithm then determines whether or not the heart sounds are coming from the same source and segments the data into two streams to represent the two sources of heartbeats. The acoustic signals are classified as either maternal or fetal using the maternal QRS positions detected by the ECG processing algorithm. If the cross-correlation of the PCG data and the maternal QRS data is high, then the PCG data stream is classified as maternal. If the cross-correlation of the PCG data with the maternal QRS is low, then cross-correlation is performed with the fetal heartbeats calculated from the ECG algorithm. If this correlation is high, the PCG data stream is classified as fetal. If neither correlation is high, the acoustic signal and the respective detected heartbeats are discarded.
The results from the independent analyses of biopotential and acoustic signals by the algorithm are grouped to extract final MHRs and FHRs by combining time-stamped annotations of the biopotential and acoustical data from detected heartbeats. Biopotential signal annotations are earlier in time than acoustic annotations of the same heartbeat. The algorithm then calculates the local variation in time differences between nearby biopotential and acoustic signal annotations and adjusts the time difference between biopotential and acoustic annotations. Missing biopotential annotations or acoustic annotations can be added if there is a corresponding signal in the other data stream in certain circumstances. The annotations are fused into one annotation per heartbeat, and HR is calculated as beats per minute.
84
Below is a diagram of our INVU algorithm for FHR and MHR.
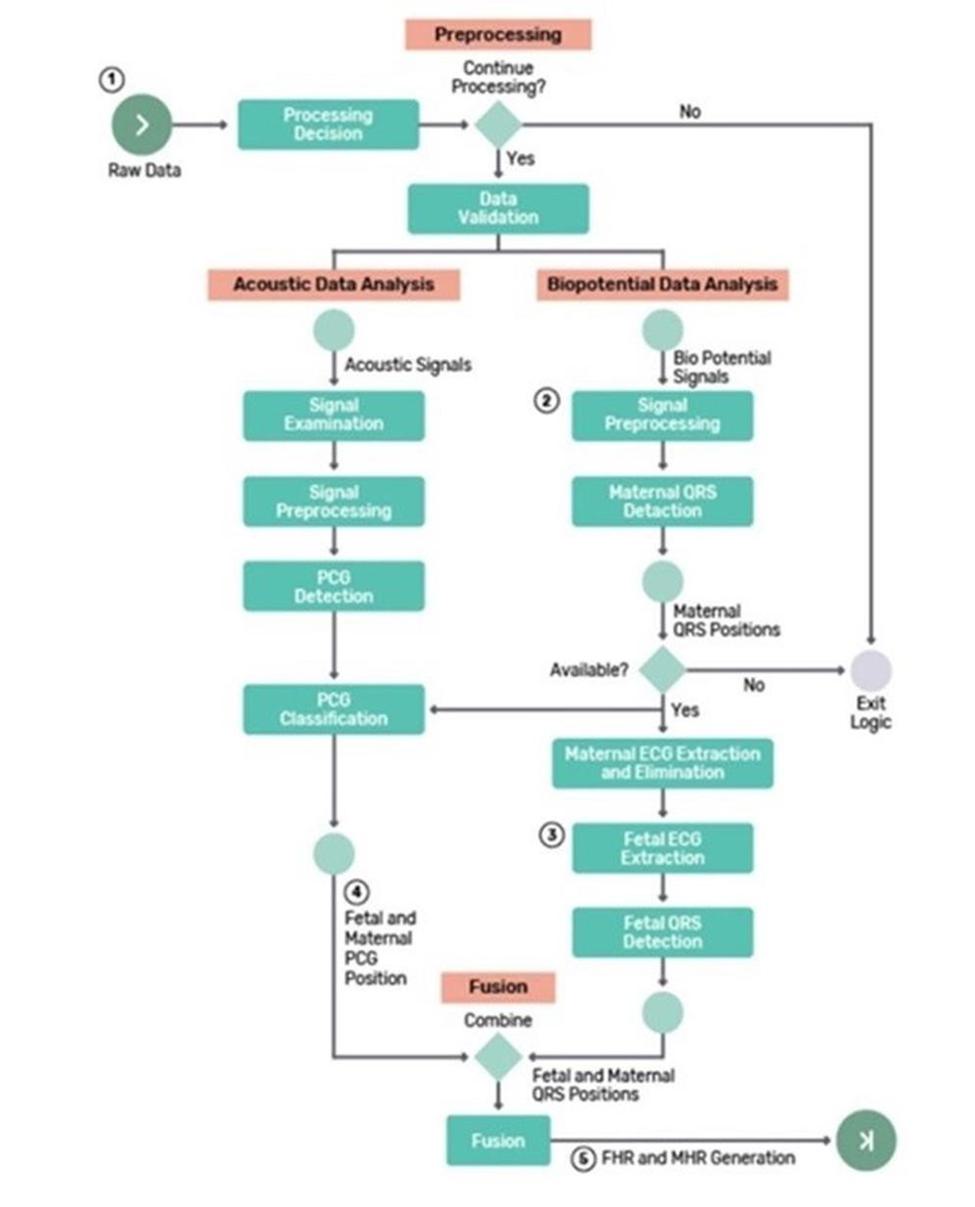
In the diagram above (1) The algorithm separately analyzes the biopotential and acoustic signals collected from our wireless sensor band in a series of signal-processing steps. (2) Signals are preprocessed to capture the relevant physiological signals and to reduce unwanted signals to noise. (3) The algorithm separates one input data stream into two groups: mECG and nonmaternal ECG. With the fECG, detailed information can be collected, including QRS waves, between waves. (4) The acoustic signals are classified as either maternal or fetal, using the maternal QRS positions detected by the ECG processing algorithm as a reference. (5) MHR and FHR measurements are generated.
85
MUA Signal Detection and Processing
We have also developed a novel algorithm that enables non-invasive, reliable MUA monitoring based on mECG and mPCG signal processing via our wireless sensor band after they pass through the uterus. Since receiving FDA clearance for MUA, and its intended use, in conjunction with MHR and FHR, for NSTs, we are now able to perform remote at home and in-clinic self-administered NSTs during the INVU monitoring period for singleton pregnancies, which we believe enhances our ability to monitor LRPs and HRPs.
The maternal heartbeat creates strong biopotential and acoustic signals, which propagate through the body and are recorded by our wireless sensor band after they have passed through the uterus. When a contraction occurs, the medium through which the signals propagate changes, causing an amplitude modulation of both the biopotential and acoustic signals in a similar manner that correlates with the mechanical effect of the contraction.
Below is a diagram of the three stages of our MUA detection algorithm.
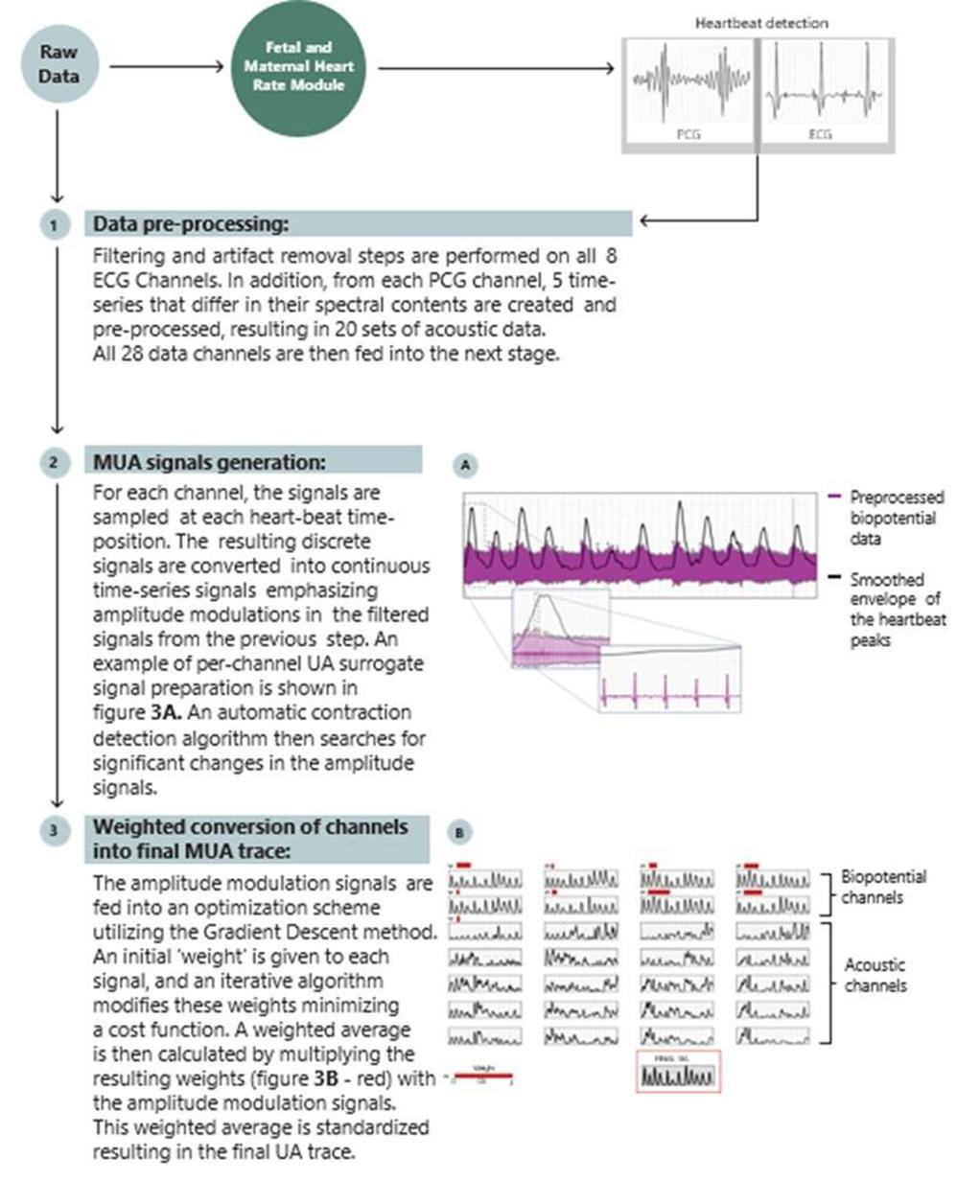
86
In the diagram above:
(a) First, data is collected from the biopotential sensors and acoustic sensors on our wireless sensor band; the raw data is preprocessed on each ECG and acoustic channel by cleaning and preparing the data for further processing.
(b) Next, all individual signals are manipulated by various computations to isolate contractions on each specific channel; the resulting signals are related to each other and iteratively merged into one final snapshot of MUA.
Cloud Computing and Software Applications
We maintain an aggregate database that captures all raw data from our wireless sensor band from all the expectant mothers. We also maintain segregated databases for each expectant mother that we build for each obstetrician, group of clinicians, academic institutions and others.
The algorithm also displays the data we are cleared to provide for visualization through one of two applications that provide tailored information for each of the clinician and expectant mother. First, a mobile application for the expectant mother, which runs on her mobile device, displays the results, controls the wireless sensor band and is utilized as a bridge to send the raw data collected to the cloud-based servers for analysis. Second, a software application for the clinician, which runs as a web-based application, our INVU Pro application, on any computer with a standard internet browser, displays a digital dashboard reflecting all expectant mothers under their care.
We believe that the application of digital tools to the data gathered by our INVU platform will enable us to identify patterns and trends that are associated with certain risks and outcomes from which we believe we will be able to make highly useful and actionable predictive recommendations to expectant mothers and their clinicians.
Clinical Studies
Approval or clearance from the FDA, or comparable regulatory agency in other jurisdictions, to capture certain measurements and perform certain tests in our clinical studies, is not guaranteed and may take longer than planned. Also, regulatory approval in one jurisdiction does not mean that we will succeed in obtaining regulatory approval in other jurisdictions.
We have completed six clinical trials and several human factor validation studies. In the first pivotal study, we clinically validated the ability to capture FHR and MHR through our wireless sensor band. Both measures were recorded simultaneously through our wireless sensor band and the current standard of care, CTG. This study successfully validated our FHR and MHR measurements as being comparable to CTG. The second pivotal study was performed to validate our ability to extract MUA. MUA was simultaneously recorded by an IUPC, TOCO and our wireless sensor band. The recorded data was then marked for contractions by three trained and board-certified maternal-fetal medicine doctors, or MFMs. Our MUA measurements were validated by direct comparison to IUPC and indirect comparison to TOCO in terms of the accuracy of capturing contractions by measuring positive agreement (“PA”) and false positive (“FP”) rate. The third clinical study was performed to evaluate the clinical utility of FHR recordings captured by our INVU platform in real world remote environments. Following a brief training session, expectant mothers recorded multiple sessions of FHR at home, after self-administering our wireless sensor band. The expectant mothers’ obstetrician then assessed the recorded data as clinically interpretable, or useful, or not. This study successfully validated the ability of our INVU platform to capture remote FHR data in real world conditions following self-administration by the expectant mothers.
Three additional studies validated our ability to successfully record remote NSTs in low and high-risk patients. A fourth clinical study to validate our ability to replace in-clinic NSTs with remote NSTs has recently been completed at UPenn. A publication titled “Utilization of a wireless monitoring device to perform nonstress tests in high-risk pregnancies from home”, summarizing the study was published in the American Journal of Obstetrics and Gynecology in June 2023. Similar to the previous study, following a brief training, expectant mothers performed prescribed NSTs at their home based on their clinical need. The expectant mothers’ obstetrician then determined whether the NST test was clinically useful. A fifth clinical study with a similar design to that of the UPenn study also aimed to assess the clinical utility of remote NSTs as well as the system usability and users’ satisfaction in high-risk patients at the Utah Valley Maternal Fetal Medicine clinic, Utah, USA. Both studies validated that NSTs may be performed at home without the need to go to the clinic. A sixth clinical study was an investigator-initiated study, designed and conducted by Sheba Hospital, Ob/Gyn department team, and aimed to assess the feasibility, logistics and patient satisfaction of hybrid care for women with gestational diabetes mellitus (“GDM”). On top of remote monitoring with INVU, each remote visit included three additional remote modalities. The results of this study demonstrated for the first time that in addition to the successful recording of remote NSTs throughout the course of the study, a hybrid maternal-fetal program for high-risk pregnancies, consisting of alternating in-person and virtual visits is feasible, saves time and improves patient satisfaction.
A detailed description of each clinical trial is provided below, followed by a table summarizing the quantitative results of all clinical studies.
87
Study 1: MHR/FHR Study
In our first pivotal clinical study, we conducted a prospective, open-label, multicenter study to compare MHR and FHR monitoring data obtained and processed by our INVU platform to CTG, the current standard of care, commencing in February 2018. The MHR/FHR study evaluated concurrent use of our INVU platform and CTG in 147 pregnant women between the ages of 18 to 50 years, with singleton pregnancies of 32 or more weeks’ gestation. The study demonstrated that the FHR and MHR outputs wirelessly obtained and processed by our INVU platform were substantially equivalent to those obtained by CTG. The study was conducted at UPenn, Eastern Virginia Medical School, Hadassah Hospital and Heidelberg University Women’s Hospital.
MHR and FHR are important measures of the expectant mother and unborn baby’s health. Various factors can affect the expectant mother or baby’s HR. These include anxiety, obesity, position and other factors. MHR can also affect FHR. A high FHR could mean that the unborn baby is not getting enough oxygen or other problems. A low FHR is often associated with low birth weight and potential fetal development issues.
Study 2: MUA Study
In our second pivotal clinical trial, we examined amplitude-modulation of mECG and PCG as a novel method for wireless non-invasive uterine monitoring to support our submission of our INVU platform for MHR and FHR for clearance to the FDA. This study evaluated the capability of our INVU platform to detect MUA during labor in comparison to IUPC. In addition, we also performed a comparison between TOCO and IUPC. An abnormal number of uterine contractions could lead to pre-term birth or impaired oxygenation for the unborn baby.
The study was a prospective, comparative, open label, multicenter study commencing in March 2019. Study sites were UPenn and an academic medical institution located in the South Central United States. The study involved 120 laboring women, 40 in the training stage and 80 in the validation stage (see details below), with a gestational age greater than 32 weeks and a BMI less than 50kg/m2 who were simultaneously monitored in a healthcare facility for 30-60 minutes with IUPC and our INVU platform. 49 of the subjects were also monitored with TOCO.
Three blinded assessors reviewed the MUA recordings obtained from our INVU platform, IUPC and TOCO and documented the presence of each contraction they identified. MUA as measured by IUPC served as the reference. PA was calculated as the percentage of IUPC contractions that were also detected on our INVU platform or TOCO tracings, within a window of +/− 30 seconds. FPs represented contractions noted with our INVU platform or TOCO, but not identified via the IUPC.
The study was divided into two separate stages – a training stage and a validation stage. In the training stage, data from 40 patients was collected from the blind assessors. The data was then unblinded and used to refine the MUA algorithm. In parallel, data collection for the validation began, but the data was not exposed and remained blinded. For the validation stage, data from 80 patients was collected. Once the training stage and the collection of data for the validation stage were completed, the algorithm was locked, and the analysis of the validation stage commenced. The analysis was performed using the locked algorithm on blinded data. The results were then passed to the statistician (external to Nuvo) which unblinded the data and analyzed the results. Nuvo remained blinded to the data until the analysis was complete.
The chart below depicts positive agreement (PA) rates and false positive (FP) rates per assessor for our INVU platform and TOCO in the training and validation stages. PA and FP rates are defined by comparing blind assessor markings of either INVU or TOCO to the blind assessor markings of IUPC. Each bar represents the PA or FP rate for a single blind assessor. Dashed lines represent the average of three assessors. Error bars show the 95% CI. In the training stage (Panel A), the PA rate is significantly higher for our INVU platform compared to TOCO (P=0.002), while the FP rate for TOCO was not significantly different from our INVU platform (P=0.06).
The results were similar to the validation phase, depicted in Panel B below. The PA rate is significantly higher for our INVU platform compared to TOCO (P<0.0001), while the FP rate for TOCO was significantly lower than our INVU platform (P<0.0001). The results indicate that MUA monitoring via our INVU platform proved to be accurate and more precise than TOCO; however, it also demonstrated a higher rate of FP compared to TOCO.
88
PA and FP Analysis
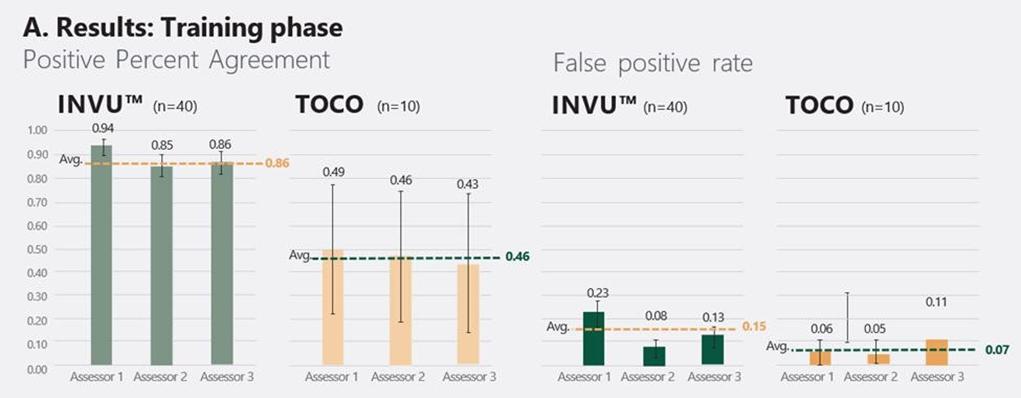
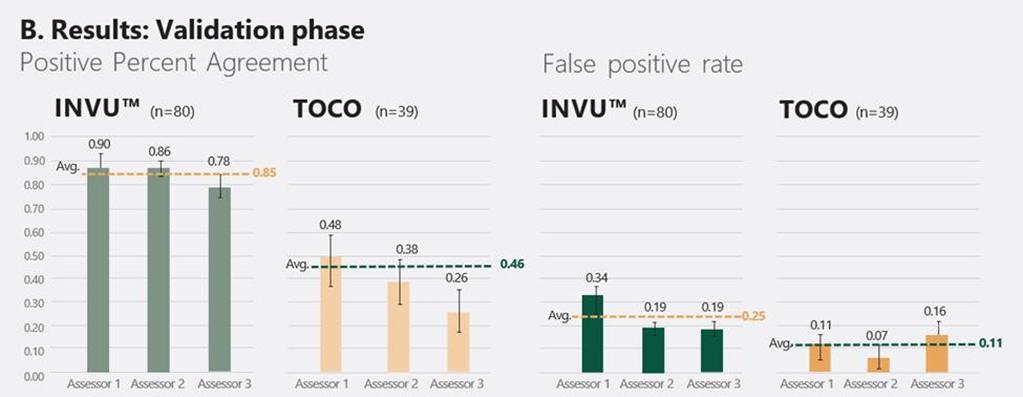
The effect of BMI on the accuracy of the INVU platform as compared to TOCO was also analyzed (see chart below). The validation data was segregated into three BMI groups: Normal (BMI < 25 kg/m2), Overweight (25≤ BMI < 30 kg/m2) and Obese (BMI ≥ 30 kg/m2). The PA rate for our INVU platform remained stable across BMI groups, while for TOCO the PA rate significantly deteriorated with increased BMI: the PA rate of TOCO was significantly lower in the obese group compared to the normal group (P=0.02). The FP rate of the INVU platform was not impacted by BMI.
89
BMI Analysis

As described above, our INVU platform detected significantly more contractions than IUPC detected. Our MUA measurements capture modulation in signals, caused by changes in the physical medium through which the signals propagate. Some of these changes cause structural changes in the uterus, and thus a modulation in the INVU signal, but do not necessarily lead to a contraction caught by the IUPC. Similar results were reported with recordings of electrohysterogram (“EHG”), which captured certain electrical activity (“EMG activity”) in the uterus that was not observed in IUPC. This excessive activity may be local electrical activity patterns, that do not amount to a full contraction and are therefore not detected by the IUPC which is pressure-dependent. Additionally, 85% of our FPs were one minute or less away from a true positive contraction. This proximity to a true positive indicates that our FPs are most likely sub-threshold activity of the uterus not captured by IUPC.
MUA monitoring via our INVU platform demonstrated a high sensitivity compared to IUPC, which exceeds that of the current standard of care. Unlike TOCO, the sensitivity of our INVU platform is not affected by maternal obesity. The high rate of FPs with our INVU platform may reflect the unique physiologic information captured within our INVU platform. This novel method for MUA detection via our INVU platform expands its remote pregnancy monitoring capabilities to include surveillance such as NSTs, which would be of great benefit to women and clinicians seeking remote solutions for HRP care. Such a study is in process with UPenn.
90
Study 3: Remote Monitoring – Self Administration
In our third clinical study, we conducted a prospective, open-label, proof of concept study with Axia and Henry Ford Health System, or HFHS, evaluating the data utility and usability of our INVU platform on pregnant subjects during virtual care prenatal visits of heart tones and blood pressure. The study commenced in April 2020 and was completed in September 2020. The study demonstrated that pregnant women can successfully use a fully remote, wireless, FHR monitoring platform to collect routine data for prenatal care.
The study evaluated pregnant women with singleton gestation of 20 or more weeks of pregnancy between the ages of 18 and 50. Ultimately, 17 women were to be evaluated by HFHS and Axia, 13 of which completed the study.
The primary objective of this research was to assess the remote clinical utility of the data output retrieved from our INVU platform. The primary performance endpoint was to evaluate if the data collected was deemed acceptable for clinical utility by clinical personnel.
The subjects underwent continuous recording of FHR and MHR for up to ten minutes per session for five days using our wireless sensor band. In addition, blood pressure was recorded using a separate third-party blood pressure monitor. All data was self-collected by each expectant mother in her home through our mobile application. The data was assessed offline by clinical personnel. No AEs were reported.
In summary, the overall percent of acceptable FHR measurements was 97.85%, which was significantly higher than the acceptance criterion of 90%. The overall percentage of acceptable blood pressure measurements was 100.0%, which was significantly higher than the acceptance criterion of 90%. The study met its prespecified goals and was deemed successful. The study demonstrated that pregnant women can successfully use a fully remote, wireless, FHR monitoring platform to collect routine data for prenatal care.
Remote NST Validation Studies – Real World Experience
Three single-center remote NST validation studies were conducted with INVU. Two of the studies were performed in the United States and the other, an investigator-initiated study, in Israel. Below is a summary of the goals and the main findings of these studies:
Study 4: UPenn (US) Study
Study Title: A Clinical Study Evaluating the Data for Remote NST Acceptability for Clinical Utility and Usability of INVU on Pregnant Subjects during Virtual Surveillance Sessions of FHR, MHR, MUA and Blood Pressure Readings.
Study Design: This was a single-center, prospective, open-label study, aimed to assess the remote clinical utility of remote fetal surveillance (NST) data output retrieved from INVU. The study was conducted at UPenn.
In addition to the evaluation of remote NST data acceptability for clinical utility by clinical personnel as the primary performance endpoint, the study aimed to evaluate the INVU system usability by the lay-user study subjects, via a standardized System Usability Scale (“SUS”) survey, as the secondary endpoint.
Following enrollment and screening procedures which included an in-clinic training with the study device, pregnant women with ≥32-week high-risk singleton gestation, or pregnancy with one child, performed self-administered fetal surveillance using the INVU device at home, one to two times per week, replacing in-clinic antepartum fetal surveillance monitoring appointments (NSTs). Monitoring data was collected by each subject in their home, through the INVU patient-facing mobile application. The data (traces) was viewed and interpreted by the clinical teams via the web-based app, the INVU Pro, for analyzing the clinical utility of data interpretability as the primary performance endpoint and reactivity, or clinical assessment of the data. Nuvo set the success criterion of this study to be achieving a rate of at least 75% remote NSTs acceptable for clinical utility.
Upon the completion of the prescribed recording sessions (the care plan), each subject was asked to complete a short usability survey. The study was completed in August 2022.
91
Results: In summary, 34 high-risk patients at mean gestational age 34.5±1.1 weeks and BMI 35.8± 6.3 kg/m2 were consented. Fetal testing indications included advanced maternal age (n=14), BMI≥40kg/m2 (n=11), gestational diabetes (n=10), chronic hypertension (n=3), antiphospholipid syndrome (n=2), and other (n=9). “n” refers to the sample size.
Five consented patients failed to complete device training. The remaining 29 patients had 131 qualifying remote NST sessions. INVU successfully obtained an interpretable NST in 93.9% (n=123) of appointments, of which 98.3% (n=121) were deemed reactive. Study subjects avoided an in-office visit 88.5% of the time. Only two appointments (1.5%) resulted in a recommendation for non-urgent delivery, both for elevated blood pressure and neither related to the NST tracing. Only one patient (2.9%) experienced mild, transient soreness at the sensor site without redness or bruising.
Twenty-three patients (79.3%) who attempted at least one remote NST completed the SUS, with a mean score of 76.5 (± 15.9) out of 100, indicating “good” usability, with 22 (95.7%) agreeing they would prefer remote NSTs over in-office testing in a future pregnancy.
Conclusions: Given the success criterion of achieving a rate of at least 75% remote NSTs acceptable for clinical utility (interpretability), the study met this endpoint as >90% were acceptable for clinical utility remote NSTs. In addition, the mean SUS score was 76.5 (+/- 15.9) out of 100 at study completion, which was indicative of good product usability. Notably, 22 of the 23 patients (95.7%) who completed the survey agreed they would prefer remote NSTs using INVU over in-clinic testing in a future pregnancy.
The findings of this study were published in the AJOG in June 2023 and confirmed the clinical utility of the INVU monitoring device for remote, at-home NSTs in high-risk pregnancies as a replacement for the vast majority of in-clinic monitoring. Our population included a range of patient demographics and indications for antenatal testing, including high rates of BMI ≥40kg/m2.
Study 5: Intermountain Health Center (US) Study
Study Title: A Single Center, Open-Label, Prospective Clinical Study Evaluating the Data Utility and Usability of Remote NST Performed by INVU System.
Study Design: A single-center, prospective, open-label study, aimed to assess the remote clinical utility of remote fetal surveillance (NST) data output retrieved from INVU. The study was conducted at Utah Valley Maternal Fetal Medicine clinic, Utah, USA.
In addition to the evaluation of remote NST data acceptability for clinical utility by clinical personnel, the study aimed to assess duration of the in-clinic training with INVU device and evaluate the INVU system usability and learnability by the lay-user study subjects via a standardized SUS survey. These were the primary endpoints. The study also aimed to assess patients’ satisfaction with the INVU system using Net Performance Score (“NPS”) survey. This was the secondary endpoint.
Following enrollment and screening procedures which included an in-clinic training with the study device, pregnant women with ≥32-week high-risk singleton gestation performed self-administered fetal surveillance using the INVU device at home, one to two times per week, on top of in-clinic antepartum fetal surveillance monitoring appointments (NSTs). Monitoring data was collected by each subject in their home, through the INVU patient-facing mobile application. The data (traces) were viewed and interpreted by the clinical teams via the web-based app, the INVU Pro for interpretation of data interpretability for clinical utility (primary performance endpoint) and reactivity (clinical assessment of the data). Upon the completion of the prescribed recording sessions (the care plan), each subject was asked to complete a short usability and satisfaction survey.
Results: The study is now closed for enrollment and official study data analysis is in process. Overall, 12 subjects were consented, eight enrolled to the study (four screen failures due to technical reasons during in clinic training with the study device) with various indications for fetal surveillance such as intrauterine growth restriction, fetal anemia, GDM, history of intrauterine fetal demise, and advanced maternal age. Enrolled subjects were at various age ranges (21-42), various gestational ages (32-35.4) and various BMI (23.9-40) at enrollment. In total, 20 remote qualified appointments were performed with a success rate of 85% (17/20 interpretable NSTs). 13 out of 17 interpretable NSTs were found reactive. We refer to “reactive” as the health status of the fetus (and not with the technical performance of INVU or the quality of the information provided by the device), with “reactive” meaning healthy and “non-reactive” possibly indicating a problem. In terms of safety, no adverse events were recorded. System usability and learnability scale (using SUS validated questionnaire) resulted in a mean score of 73 (N=8), Grade: B. The mean NPS score was (1-10 scale, N=7) 8.8 (71.4 % Promoters; 14.3 % Passives; 14.3 % Detractors). Promoters are those who responded with a score of 9 or 10, Passives are those who responded with a score of 7 or 8 and Detractors are those who responded with a score of 0 to 6. The NPS score is calculated with the following formula: total % of promoters – total % of detractors = NPS.
92
Study 6: Sheba (Israel) Investigator Initiated Study
Study Title: Feasibility of Remote Care Visits for Women with Gestational Diabetes
Goal: To assess the feasibility, logistics and patient satisfaction of hybrid care for women with GDM.
Study Design: A single-center, prospective, open-label study in which women with GDM (one or two), pre-pregnancy BMI ≥15 and ≤40 and carrying a singleton pregnancy at ≥31 gestational weeks were offered to participate. The patient’s journey included in-person and remote visits taking place alternately for a duration of four weeks. The remote visit included maternal assessment – vital signs, glycemic control (documented in Datos app), urinalysis (Healthy.io), FHR assessment (INVU) and ultrasound measured maximal vertical pocket (PulseNmore) yielding a modified biophysical profile (“mBPP”). Usefulness, ease of use, effectiveness, reliability, and patient satisfaction of the entire remote visit were assessed with the telehealth usability questionnaire (“TUQ”).
Among other tested technologies, study participants underwent a remote blood pressure measurement using the cuff provided, as well as FHR, MHR and NST monitoring using INVU. During this study, MUA trace was not presented on the INVU Pro App and an NST evaluation was based on FHR, MHR and additional clinical data as per team’s considerations, with no visibility of the MUA tracing. INVU remote sessions were conducted twice in total at weeks one and three following enrollment, during which the investigator evaluated the NST acceptability for clinical utility. NST evaluation was based on FHR, MHR and additional clinical data as per the investigator considerations. MUA was not presented. Monitoring data was collected by each subject in their home, through the INVU patient-facing mobile application. The data (traces) were viewed and interpreted by the clinical teams via INVU Pro.
Upon the completion of the prescribed recording sessions (the care plan) with INVU, the subjects completed an SUS and NPS survey.
Study Status: Closed for enrollment from July 2022.
Study Results: Overall, 22 subjects diagnosed with GDM were consented, 21 subjects passed screening procedures which included successful hands on in clinic training with the study device. Enrolled subjects were at various age ranges (24-46) and various BMI (23-41) at enrollment. Out of four weekly appointments, two were conducted remotely (every other week). In total, 39 remote appointments were performed with a success rate of 97.4% (38/39). There was also a high safety profile as no device or treatment related adverse events were reported. The system usability and learnability scale with INVU (using an SUS validated questionnaire) presented a mean score: 75.5 (N=19), Grade: B. The mean NPS score (1-10 scale) resulted in 9.4 (83.5% Promoters; 11% Passives; 5.5% Detractors). Additionally, total remote visit length was significantly shorter (65.4±21.6 min) compared to the in-person visit (171.1±21.4 min, P<0.001). The second remote visit was significantly shorter (58.7±23.5 min) than the first remote visit (73.1±29.4 min, P=0.03). Lastly, TUQ results indicated high usability of the telehealth visit overall (6.6/7).
The results of this study have been submitted for publication in The Lancet Digital Medicine, and has demonstrated that a hybrid maternal-fetal program for high-risk pregnancies, consisting of alternating in-person and virtual visits is feasible, saves time and improves patient satisfaction.
Human Factor Studies – System Usability
We also conducted several studies with FDA-recognized Human Factor facilities, Medstar Health National Center for Human Factors and CORE Human Factors, Inc. related to the ability of participants to utilize our former platform in remote settings, PregSenseTM. In total, 15 pregnant women participated in the Medstar study (“HF Validation 1 Study”), 16 pregnant women participated in the CORE study (“HF Validation 2 Study”), and 15 pregnant women participated in the second CORE study (“HF Validation 3 Study”).
In the HF Validation 1 Study, data collection and analysis focused on safety-critical tasks, with a secondary focus on error trends associated with device functionality that did not have negative safety implications (i.e., non-critical tasks). The users did not have any assistance. We believe the results of this study established that the design implementation and instructive materials for our INVU platform facilitated remote use and self-administration. Overall, participants completed four out of five testing scenarios without performing any critical, safety-related errors. We implemented a number of design modifications based on study recommendations to address concerns.
93
The HF Validation 2 Study was a supplemental study conducted following FDA feedback and was designed to assess the above mentioned, newly implemented mitigations to critical task errors observed in the HF Validation 1 Study and were found to be effective. Many non-critical tasks were assessed as part of the study. However, the following tasks are the most relevant to self-administration of the device and home monitoring:
| ● | 14/15 participants (93.3%) completed the monitoring session tasks as expected; one participant performed one of the five tasks incorrectly (non-critical error), but still initiated a successful monitoring session. |
| ● | All participants (15/15) successfully ran a monitoring session by self-administering the PregSense™ device and recording fetal heart rate, without apparent difficulty or confusion. |
| ● | For the non-critical knowledge tasks during this session, all participants were able to correctly describe what the fetal heart rate display was in the PregSense™ mobile application when the moderator pointed to it. |
| ● | Most participants (13/15) were able to name all of the data shown on the patient facing PregSense™ platform during the monitoring session; 2/15 participants had partially correct answers on this knowledge task. |
In a post-performance debrief, 14/15 participants affirmed that they could routinely use the PregSense™ platform safely and successfully if it was prescribed as part of their care plan from their healthcare provider.
Overall, results of these two validation studies demonstrated that the design of labeling and instructional materials provided multiple mitigation strategies to prevent the occurrence of these errors to a reasonable extent. The mitigations were found to be effective. We believe the results of these studies established that the design implementation and instructive materials for our remote monitoring platform facilitated remote use and self-administration.
The HF Validation 3 Study performed by Core Human Factors evaluated the INVU system components intended for use by pregnant women. The study included a total of 15 participants representing the intended user group, pregnant women in their 32nd week or later of gestation with a singleton pregnancy. Nuvo conducted a use-related risk analysis of the product and identified four critical tasks, which were all tested in a human factors validation study in January 2021. A pilot study was conducted on the product in advance of the study, which resulted in changes to the Moderator’s Guide.
All participants in the HF Validation 3 Study completed the following critical assessments successfully and established that they knew:
| ● | not to use on an opened wound or infected skin; |
| ● | not to use if allergic to band’s materials; and |
| ● | to position or restrain the power cable in a safe way avoiding strangulation. |
Two participants encountered a close call relating to the following critical assessment: Knows not to wear the band while charging. These two participants initially reported that they would charge the band while wearing it, but later referred to the Quick User Guide (“QUG”) and identified that they should not charge the band while wearing it. Additionally, participants were assessed on a task that was previously identified as critical (Knows to charge until the LED light turns green or until four hours), but following testing, Nuvo identified that there is no actual risk and there would be no harm to a user if the band was left charging for prolonged periods. Therefore, the task was removed from the use-related risk analysis and associated warnings were removed from the labeling.
Nuvo has not identified any significant residual risk that remained, and we believe product users will benefit from being able to monitor their pregnancy remotely.
94
Below is a tabulated summary of the objectives and endpoints and main results and conclusions of the clinical studies conducted with INVU:
| # | Study Title |
Goal: Objectives & |
Clinical Sites |
No. of Participants |
Main Results |
Conclusions |
| 1. |
Clinical Study Evaluating the Safety of the Non-Invasive INVU and Comparative Performance of INVU versus CTG in Prenatal Monitoring of Pregnant Subjects
|
Objective:
To assess the agreement between INVU data collection, and values measured via the standard of care used for prenatal monitoring (i.e., CTG) and to assess the safety of INVU.
Performance Measures:
FHR & MHR as measured by INVU vs SOC (i.e., CTG)
Safety Measures:
The incidence of device related and/or protocol procedure related adverse events and/or serious adverse events related to the study device, INVU.
|
4 sites:
Hadassah, Israel
UPenn, USA
EVMS, USA
Heidelberg, Germany
|
147 |
Performance - FHR:
● Mean bias of INVU versus CTG FHR is -0.2974 bpm or on average, INVU was 0.2974 bpm less than CTG, which was not clinically significant (95% CI: [-0.7702, 0.1753]), and its standard deviation 4.357 bpm (95% CI: [4.327, 4.388]).
● This means that INVU underestimates the FHR by 0.2974 bpm on average. This difference has no clinical significance, see below.
● Overall accuracy is -0.2974 bpm ± 4.357 bpm, or the average level of accuracy was 0.2974 within a range of error of 4.357 bpm.
● Same as above – level of accuracy and the range of an error.
● 95% limits of agreement are [-8.84, 8.24].
● The limits of agreement represent the range within which most differences between the two measurements will lie.
● Therefore, the acceptance criterion is met for FHR.
● The acceptance criterion for FHR was set to +/ 10 bpm, since the limits of agreement for FHR were -8.84-8.24, the pre-specified performance goals of this study have been met. We conclude that FHR as measured by INVU closely resembles that of the CTG measurements.
Performance - MHR:
● Mean bias of INVU versus CTG MHR is 0.2844 bpm, or on average INVU was 0.2844 bpm more than CTG, which was not clinically significant (95% CI: [0.2419, 0.3269]), and its standard deviation 2.847 bpm (95% CI: [2.829, 2.865]).
● Overall accuracy is 0.2844 bpm ± 2.847 bpm.
● This means that INVU overestimates the MHR by 0.2844 bpm on average. This difference has no clinical significance. See below.
● 95% limits of agreement are [-5.30, 5.86] bpm.
● The limits of agreement represent the range within which most differences between the two measurements will lie.
● Therefore, the acceptance criterion is met for MHR.
Safety:
● No Adverse Events were reported. The adverse event rate is therefore 0% (0/147). |
1. FHR and MHR as measured by INVU closely resemble that of CTG measurements.
2. We believe INVU may be used for pregnant women at gestational age ≥32 + 0 weeks with a singleton pregnancy.
|
95
| # | Study Title |
Goal: Objectives & |
Clinical Sites |
No. of Participants |
Main Results |
Conclusions |
| 2. | Clinical Study Evaluating the Safety of INVU and Comparative Performance of INVU versus IUPC in Prenatal Monitoring of Pregnant Subjects with Uterine Contractions |
Objective:
To assess the agreement between INVU and IUPC during prenatal monitoring of uterine contractions and to assess the safety of INVU. Additional information comparing to TOCO (the standard of care) was also collected in the CLP2001 sub-study.
Primary Performance Measures:
Uterine contractions as collected via INVU system compared to the data collected via IUPC monitoring system. In the sub-study, comparison to TOCO was also performed, in addition to IUPC.
Safety Measures:
All adverse events and serious adverse events related and unrelated to INVU use. |
2 sites:
1. UAMS, USA
2. UPenn, USA
|
78 |
Performance:
● Positive agreement rate between IUPC and INVU for all three assessors together along with the 95% CI of the positive agreement rate is 84.80% (95% CI: [81.58%; 88.02%]).
● One can be 95% certain that the real positive agreement rate (i.e., in the full population and not in this sample) between INVU and IUPC is between 81.58% and 88.02%.
● The overall positive agreement was higher with the INVU device than with TOCO, which demonstrated a positive agreement rate of 37.50% (95% CI: [28.23%; 46.77%]).
● One can be 95% certain that the real positive agreement rate (i.e., in the full population and not in this sample) between TOCO and IUPC is between 28.23% and 46.77%.
● No statistically significant differences were observed in positive agreement between the BMI groups (pairwise p-values >0.1331) when using the INVU device. However, the positive agreement of the TOCO was higher in the BMI<25 group versus the BMI ≥30 group (p-value: 0.0244).
● We cannot say that the positive agreement rate is different between the BMI groups when using the INVU, but when using TOCO, the rate was higher in the BMI<25 group compared to the BMI ≥30 group.
● The overall false positive rate in the number of contractions identified compared to IUPC was higher with the INVU device: 24.8% (95% CI: [21.97%; 27.60%] than with TOCO (10.7%) (95% CI: [5.65%; 15.72%]).
● The false positive rate (No. of contractions identified by INVU/TOCO and that are not present in the IUPC trace) with INVU is 24.8% and with TOCO is 10.7 %. INVU is more sensitive than TOCO.
● Clinical Interpretability: Overall, the IUPC was considered interpretable or partially interpretable in 100.0% of the cases (120/120), INVU in 97.5% (117/120), and TOCO in 92.5% (111/120) of the cases.
● Session interpretability=session that can be interpreted by a clinical team and used for clinical diagnosis.
● The pre-specified performance goals of this study were met.
Safety:
● No device-related adverse events were observed in the validation study. A single non-device related adverse event was reported. |
1. INVU performance has met this goal by presenting a positive agreement of 84.80% (95% CI: [81.58%; 88.02%]) compared to IUPC.
The study has met its prespecified goals and is deemed successful.
2. INVU outperforms the standard of care, TOCO, in positive percent agreement.
3. INVU demonstrates similar accuracy across BMI ranges. By comparison, for TOCO, the positive agreement of the obese group (BMI≥30) was significantly lower than that of the normal group (BMI<25).
4. The reported false positive rate in the INVU device was consistent with published literature for TOCO, although higher than in the TOCO group in the present study.
5. There were no safety issues associated with use of the device.
|
96
| # | Study Title |
Goal: Objectives & |
Clinical Sites |
No. of Participants |
Main Results |
Conclusions |
| 3. | A Clinical Study Evaluating the Data Utility and Usability of INVU on Pregnant Subjects during Virtual Care Prenatal Visit, of Heart Tones and Blood Pressure |
Objective:
To assess the remote clinical utility of the data output retrieved from INVU.
Performance Measures:
FHR and blood pressure data retrieved from the INVU system are acceptable for clinical utility by clinical personnel.
Safety Measures:
The incidence of device related and/or protocol procedure related adverse events and/or serious adverse events related to the study device, INVU. |
2 sites:
1. Axia Women’s Health, USA
2. Henry Ford Medical Group, USA |
13 |
Performance:
● The overall percent of acceptable FHR measurements is 97.85% (Exact binomial two-sided 95% CI: [92.52%; 99.74%]), the lower confidence bound is 92.52%.
● One can be 95% certain that the percent of acceptable FHR measurements (i.e., in the full population and not in this sample) is between 92.52% and 99.74%, i.e., > 92.52%. To evaluate whether the study was successful and met its prespecified success criterion, this last value should be compared to the acceptance criterion.
● It is significantly higher than the acceptance criterion of 90%.
● The overall percent of acceptable blood pressure measurements was 100.0% (Exact binomial two-sided 95% CI: [96.11%; 100.0%]), the lower confidence bound is 96.11%.
● One can be 95% certain that the percent of acceptable blood pressure measurements (i.e., in the full population and not in this sample) is between 96.11% and 100.0%, i.e., > 96.11%. This last value should be compared to the acceptance criterion.
● It is significantly higher than the acceptance criterion of 90%.
Safety:
● No Adverse Events were reported. The adverse event rate is therefore 0% (0/13). |
1. The study has met its prespecified goals and is deemed successful.
2. The INVU platform provided clinically acceptable FHR data and blood pressure measurements in almost all recording sessions from the wireless, remote, self-administered monitoring platform used by the pregnant woman at home.
3. We believe INVU can be used in pregnant women at gestational age ≥20 + 0 weeks with a singleton pregnancy. |
97
| # | Study Title |
Goal: Objectives & |
Clinical Sites |
No. of Participants |
Main Results |
Conclusions |
| 4 |
UPenn Study
A Clinical Study Evaluating the Data for Remote Non-Stress Test Acceptability for Clinical Utility and Usability of INVU on Pregnant Subjects during Virtual Surveillance Sessions of FHR, MHR, MUA and Blood Pressure Readings |
Objective:
To assess the clinical utility of remote NST data output retrieved from INVU.
Performance Measures:
● Primary - Evaluation of remote NST acceptability for clinical utility by clinical personnel.
● Secondary-Evaluation of the INVU system usability by the lay users.
● Exploratory-
- NST Assessment (reactive/non-reactive) – evaluation of NST reactivity based on remote NST data output.
- Evaluate whether participants required an additional in-clinic assessment.
Safety Measures:
All adverse events including serious adverse events related and unrelated to INVU use. |
1 site:
UPenn, PA USA
|
29 |
Performance:
● A total of 34 subjects were enrolled in the entire study. Of them, first ten subjects were enrolled for INVU fetal monitoring without MUA and additional 24 subjects were enrolled for the complete NST recording, including MUA.
● 34 subjects consented, 29 enrolled (five failed to complete device training).
● Out of 131 remote NST sessions, 123 were ‘clinically interpretable’ (93.9%).
● 121 out of 123 interpretable sessions were deemed reactive (reactive NST) (98.3%).
● Study subjects avoided an in-office visit 88.5% of the time.
● Only two (1.5%) appointments resulted in a recommendation for non-urgent delivery, both for elevated blood pressure and neither related to the NST tracing.
● Twenty-three patients (79.3%) who attempted at least one remote NST completed the SUS, with a mean score of 76.5 (± 15.9) out of 100, indicating “good” usability, with 22 (95.7%) agreeing they would prefer remote NSTs over in-office testing in a future pregnancy.
Safety:
● One study participant (2.9%) experienced mild soreness at the sensor site without redness or bruising.
● Six out of eight Adverse Events took place in the MUA cohort, but none was related to the device, or the study procedure. |
● Given the success criterion of achieving a rate of at least 75% remote NSTs acceptable for clinical utility (interpretability), the study has met this goal by presenting >90% acceptable for clinical utility remote NSTs.
● In addition, the mean SUS score was 76.5 (+/- 15.9) out of 100 at study completion, which was indicative of good product usability. Notably, 22 of the 23 patients (95.7%) who completed the survey agreed they would prefer remote NSTs using INVU over in-clinic testing in a future pregnancy.
● The findings of this study confirm the clinical utility of INVU monitoring device for remote, at-home NSTs in high-risk pregnancies as a replacement for the vast majority of in-clinic monitoring.
● Our population included a range of patient demographics and indications for antenatal testing, including high rates of BMI ≥40kg/m2 and presented a high safety profile. |
98
| # | Study Title |
Goal: Objectives & |
Clinical Sites |
No. of Participants |
Main Results |
Conclusions |
| 5 |
Intermountain Study (Utah)
A Single Center, Open-Label, Prospective Clinical Study Evaluating the Data Utility and Usability of Remote NST Performed by INVU System
|
Objective:
The primary objective of this research is to assess the clinical acceptability of remote NST retrieved from INVU.
Performance Measures:
Primary –
● To evaluate remote NST acceptability for clinical utility by clinical personnel.
● To assess duration of the in-clinic training with INVU device.
● To evaluate INVU system usability and learnability by the lay users.
Secondary –
● To assess patients’ satisfaction with INVU system.
Safety Measures:
● All adverse events including serious adverse events related to INVU use. |
1 site:
Utah Valley Maternal Fetal Medicine clinic, USA
|
8 |
Performance:
● 12 subjects were consented, eight enrolled to the study (four screen failures due to technical reasons during in clinic training with the study device) with various indications for fetal surveillance such as IUGR, fetal anemia, GDM, history of intrauterine fetal demise, adverse maternal age, etc.
● 20 remote qualified appointments were performed with a success rate of 85% (17/20 interpretable NSTs).
● 13 out of 17 interpretable NSTs were found reactive.
● System usability and learnability scale (using SUS validated questionnaire) – Mean score: 73 (N=8), Grade: B.
● Mean NPS score (1-10 scale, N=7): 8.8 (71.4 % Promoters; 14.3 % Passives; 14.3 % Detractors).
Safety:
● No Adverse Events were recorded. |
This study confirms the clinical utility of INVU for remote, at-home NSTs in high-risk pregnancies.
*Official Clinical Study Report is in process.
|
99
| # | Study Title |
Goal: Objectives & |
Clinical Sites |
No. of Participants |
Main Results |
Conclusions |
| 6 |
Sheba IIS Study
Feasibility of Remote Care Visits for Women with Gestational Diabetes
|
Primary Research Question:
Is a paradigm shift from in-person clinical visits to a hybrid model of in-person and remote visits feasible among the population of women with GDM?
Secondary Research Questions:
Secondary research questions the study will address are:
● Is it clinically feasible to replace each of the following components of in-person maternal assessment (anamnesis, vital signs, urine test strip, glycemic control assessment) with remote assessment?
● Is it clinically feasible to replace each of the following components of in-person fetal assessment (monitor and ultrasound maximal vertical pocket) with remote assessment?
● Do remote visits improve patient satisfaction compared to an in-person visit (in general and individually for each component of maternal assessment)?
● Are remote visits shorter compared to the duration of an in-person visit (in general and individually for each component of fetal assessment)? |
1 site:
The Josef Buchmann Gynecology and Maternity Center (Ob/Gyn)
Sheba Medical Center
|
21 |
Study results (INVU-related):
● Overall, 22 subjects diagnosed with GDM were consented, 21 subjects passed screening procedures which include successful hands on in clinic training with the study device. Enrolled subjects were at various age ranges (24-46) and various BMI (23-41) at enrollment. Out of four weekly appointments, two were conducted remotely (every other week).
● In total, 39 remote appointments were performed with a success rate of 97.4% (38/39).
● High safety profile - no device or treatment related adverse events were reported.
● System usability and learnability scale with INVU (using SUS validated questionnaire) – Mean score: 75.5 (N=19), Grade: B.
● Mean NPS score (1-10 scale): 9.4 (83.5% Promoters; 11% Passives; 5.5% Detractors).
● Total remote visit length was significantly shorter (65.4±21.6 min) compared to the in-person visit (171.1±21.4 min, P<0.001). The second remote visit was significantly shorter (58.7±23.5 min) than the first remote visit (73.1±29.4 min, P=0.03).
● TUQ results indicate high usability of the telehealth visit overall (6.6/7). |
The results of this study had demonstrated that a hybrid maternal-fetal program for high-risk pregnancies, consisting of alternating in-person and virtual visits is feasible, saves time and improves patient satisfaction.
* Official Clinical Study Report is in process. |
100
Below is a tabulated summary of the participant enrollment, determination of the value of success, additional administration and reporting information, and parameters of the usability scale and its relation to related datapoints of, the clinical studies conducted with INVU:
| Study | Question | Answer | Ref |
|
Study 1: MHR/FHR Study
|
Why did particular participants not complete enrollment or drop out of study? |
Out of 151 screened subjects, two subjects were not enrolled: one was a screening failure (pregnant with twins) and the other one withdrew consent.
Out of 149 enrolled to the study subjects, two subjects dropped out due to device failure (data was not recorded due to connectivity issues). |
CLINICAL STUDY CLI1000 REPORT (Ref. No.: REP00229)
PIVOTAL CLP1000 STATISTICAL ANALYSIS REPORT
(Ref. No.: REP00213) |
| How did Nuvo determine the value for success of the study? |
The success criterion for the study was determined as follows: “The study will be deemed successful if the limits of agreement (LOA’s) for FHR are within the interval [-10, 10] Bpm and MHR between INVU and the standard of care device are within the interval [-7, 7] Bpm”.
This success value was based on the data from the literature, where the limits of agreement for both FHR and MHR with FSE, abdominal Fetal ECG and ultrasound were clinically evaluated showing that the abdominal fetal ECG limits of agreement are (± 1.96 SD) 8.40; −8.729. | ||
| Who administered and reported the study? |
The study was conducted in agreement with the current International Conference on Harmonization (ICH) guidelines on Good Clinical Practice (GCP).
The study management and reporting were conducted by Nuvo. | ||
| Was the same system usability scale used for each study? | N/A | ||
| How were the usability figures or success of the study calculated from the SUS scores? | N/A | ||
| How did the patient’s BMI factor into successful use of the device? |
The pre-specified goals of this study have been met and the study results demonstrated high correlation between INVU and CTG in patients’ population with prior to pregnancy BMI range from 16.5 to 44.9 kg/m2. | ||
101
| Study | Question | Answer | Ref |
|
Study 2: MUA Study
|
Why did particular participants not complete enrollment or drop out of study? |
The study was divided into Training and Validation stages.
In the Training stage – all the screened subjects, 44, completed screening and were enrolled. Out of 44 enrolled, four subjects were withdrawn (did not complete all the study procedures). Two out of four withdrew due to technical reasons (one- no data was displayed despite four attempts with two different belts; two- low battery in the tablet presenting the user interface and monitoring data, therefore the session could not be conducted).
Additional two subjects were withdrawn due to clinical reasons. One subject withdrew consent prior to the beginning of the monitoring session and the other subject had a medical issue after consenting but prior to beginning of monitoring session. No study-related procedures were done, and no data was recorded. |
Clinical Study CLI2000 report (Ref. No.: REP00565)
Statistical report MUA validation CLP2000 CLP2001_Final 1.0 |
| How did Nuvo determine the value for success of the study? |
Based on review of published literature regarding the standard of care, tocodynamometry (TOCO) and its performance relative to IUPC, the study was to be deemed successful if the percent of positive agreement between IUPC and INVU™ in identifying a contraction is at least 85% (point estimate). Because FDA typically requires a performance goal to be defined in terms of confidence limits, and not only point estimates, the success criterion in the protocol was subsequently described in the Statistical Analysis Plan as a formal statistical hypothesis for which the success criterion based on the comparison to the lower confidence limit, but without changing the level of performance required for success.
The null hypothesis was to be rejected in favor of the alternative hypothesis and the study deemed successful if the lower limit of the confidence interval is greater than 75%. |
102
| Study | Question | Answer | Ref |
| Who administered and reported the study? | The study was sponsored and managed by Nuvo in compliance with the applicable, local and international guidelines. | ||
| Was the same system usability scale used for each study? | N/A | ||
| How were the usability figures or success of the study calculated from the SUS scores? | N/A | ||
| How did the patient’s BMI factor into successful use of the device? |
The BMI covers a broad and representative range of patients. The prior to pregnancy BMI range was [17.2-47.5] kg/m2.
No statistically significant difference was observed between the different BMI groups as opposed to a significant impact of BMI in TOCO group, where a positive agreement of the obese group (BMI≥30) was significantly lower than that of the normal group (BMI<25), in line with literature demonstrating a reduction of sensitivity in TOCO compared to IUPC, with increased BMI (Refs 12, 17 and 22 in study report). |
||
103
| Study | Question | Answer | Ref |
|
Study 3: Remote Monitoring – Self Administration
|
Why did particular participants not complete enrollment or drop out of study? | Out of 17 screened subjects, four were screening failures due to a failure to complete the training session with a demo kit. | |
| How did Nuvo determine the value for success of the study? | The primary performance measure was the evaluation of the remotely recorded FHR and MHR and their acceptance for clinical utility. The study success criterion was set at the rate of at least 90% of acceptable for clinical utility FHR signal. This goal was based on Nuvo’s internal development and commercial strategies and on discussions with physicians on what will be an acceptable success rate. | ||
| Who administered and reported the study? | The study was sponsored and managed by Nuvo in compliance with the applicable, local and international guidelines. | ||
| Was the same system usability sale used for each study? | No usability scale was applied. | ||
| How were the usability figures or success of the study calculated from the SUS scores? | N/A | ||
| How did the patient’s BMI factor into successful use of the device? | The prior to pregnancy BMI range was [20.08-48.0] kg/m2. The BMI covers a broad and representative range of patients that did not affect the success rate of the study. | ||
104
| Study | Question | Answer | Ref |
| Study 4: UPenn (US) Study | Why did particular participants not complete enrollment or drop out of study? |
Five out of 34 subjects did not complete screening procedures (screening failures) due to a failure to achieve continuous tracing during in-clinic training session.
Nine out of 29 enrolled and remotely monitored subjects withdrew during the study conduct: two – due to clinical reasons (no longer has NST indication, elevated blood pressure during last session – non-related to the study device), six withdrew consent and one – lack of compliance with study procedures. |
Clinical Study CL8000 report
Ref. No.: REP00740 |
| How did Nuvo determine the value for success of the study? | The expected NST data acceptability for clinical utility success rate was set at 75%. The value of the success criterion was based on Nuvo’s developmental and commercial goals and on discussions with physicians on what will be clinically expectable. | ||
| Who administered and reported the study? | The study was sponsored and managed by Nuvo in compliance with the applicable, local and international guidelines. | ||
| Was the same system usability scale used for each study? | The same usability score was used for this study. |
105
| Study | Question | Answer | Ref | |
| How were the usability figures or success of the study calculated from the SUS scores? | ● | Upon the completion of the prescribed recording sessions (the care plan), each subject was asked to complete a short system usability survey (SUS). | ||
| ● |
SUS is a widely used for assessing the usability of a system, product, or service.
It provides a standardized and quantifiable measure of perceived usability.
The SUS questionnaire consists of a set of 10 statements or items related to the usability of the system being evaluated. Participants respond to each statement using a five-point Likert scale, ranging from “Strongly Disagree” to “Strongly Agree.” The scale is then scored and transformed to provide a usability score. |
|||
| ● | For the calculation of SUS score, there is a formula that involves summing the scores, multiplying by a constant, and making adjustments (SUS Score = 2.5*(20 + sum (odd questions)-sum (even questions) | |||
| ● | The resulting score is on a scale from 0 to 100, where higher scores indicate better usability. | |||
| ● |
The total score of the usability survey was calculated per subject. Then, the one-sided 95% confidence interval of the average score for all subjects was calculated. |
|||
| ● | Out of the overall study PA dataset (29), the number of study subjects who completed the SUS questionnaire is 23. The mean Usability Score is 76.5 (SD:15.9). | |||
106
| How did the patient’s BMI factor into successful use of the device? |
The prior to pregnancy BMI range was [20.8-44.3] kg/m2; BMI range at enrollment was [25.1-47.8] kg/m2;.
Out of 34 subjects, 11 subjects were indicated for NST due to high BMI (BMI>40 kg/m2).
Given the success criterion of achieving a rate of at least 75% remote NSTs acceptable for clinical utility (interpretability), the study has met this goal by presenting >90% acceptable for clinical utility remote NSTs.
The findings of this study confirm the clinical utility of the INVU monitoring device for remote, at-home NSTs in high-risk pregnancies as a replacement for the vast majority of in-clinic monitoring. Our population included a range of patient demographics and indications for antenatal testing, including high rates of BMI ≥40kg/m2. These data have the potential for widespread clinical application. |
||
107
| Study | Question | Answer | Ref |
| Study 5: Intermountain Health Center (US) Study | Why did particular participants not complete enrollment or drop out of study? | Overall, 12 subjects were consented, eight enrolled to the study (four screen failures due to technical reasons during in clinic training with the study device). |
Study protocol
Ref: Ref. No.: TP00175 |
| How did Nuvo determine the value for success of the study? | It was a small scale, commercial pilot validation study. No success criterion was set. | ||
| Who administered and reported the study? | The study was sponsored and managed by Nuvo in compliance with the applicable, local and international guidelines. | ||
| Was the same system usability scale used for each study? | The same usability score was used for this study. | ||
| How were the usability figures or success of the study calculated from the SUS scores? |
Details on the SUS score calculation are presented above.
For this study results, SUS resulted in a mean score of 73 (N=8), Grade: B. |
||
| How did the patient’s BMI factor into successful use of the devoce? | Enrolled subjects were at various BMI (23.9-40 kg/m2) at enrollment. In total, 20 remote qualified appointments were performed with a success rate of 85% (17/20 interpretable NSTs). | ||
108
| Study | Question | Answer | Ref |
|
Study 6: Sheba (Israel) Investigator Initiated Study
|
Why did particular participants not complete enrollment or drop out of study? | 22 subjects diagnosed with GDM were consented, 21 subjects passed screening procedures. One subject was a screening failure due to a failure to complete in clinic hands on training session. | Sheba study protocol and initial data analysis |
| How did Nuvo determine the value for success of the study? |
This is an investigator-initiated feasibility study the goal of which was to assess the feasibility of hybrid care for pregnant women diagnosed with gestational diabetes. The visits (remote and in person) included testing INVU and three additional modalities.
No specific success criterion was set. |
||
| Who administered and reported the study? | Sheba Hospital | ||
| Was the same system usability scale used for each study? | The same usability score was used for this study. | ||
| How were the usability figures or success of the study calculated from the SUS scores? | The system usability scale with INVU (using an SUS validated questionnaire) presented a mean score: 75.5 (N=19), Grade: B | ||
| How did the patient’s BMI factor into successful use of the device? |
Enrolled subjects were at various BMI (23-41kg/m2) at enrollment.
In total, 39 remote appointments were performed with a success rate of 97.4% (38/39). There was also a high safety profile as no device or treatment related adverse events were reported. |
||
109
Commercial Relationships
We have a number of commercial relationships in place and are seeking to develop additional relationships in each of the five categories described below. We believe that while none of these relationships and agreements are individually material to our commercial success, however, they are an important directional signal of our confidence in our commercial strategy as a whole and provide investors with relevant information about our progress.
Commercial Customers
Commercial customers are healthcare providers with an installed base of clinicians that understand how to prescribe and use our INVU platform for the expectant mothers under their care. We acquire these commercial customers by selling directly to two primary categories of customers: (i) hospital and healthcare systems and (ii) private practices, including independent private practice and physician practice management, or PPM, groups. In addition to our current commercial customers, some of which are outlined below, we are in discussions to work with other hospitals and healthcare systems, and PPM groups with scale, size and value contracting ability.
Hospital and Healthcare Systems – We are targeting large hospital groups and healthcare systems that manage thousands of pregnancies per year. With these customers, we are primarily focused on accelerating market adoption with our Philips partnership. See “—Expanded Commercial Partnership.” Some current commercial customers in this category are:
| ● | Banner Health. Banner Health is a non-profit health system that is recognized as one of the top health systems in the country for the clinical quality consistently provided to patients in their hospitals. Banner Health is headquartered in Phoenix, Arizona, and operates 30 hospitals, including three academic medical centers and other related health entities and services in six states. Nuvo has completed the first phase of a commercial pilot agreement, that was entered into in September 2022, with Banner Health that successfully leveraged INVU towards increasing operational efficiency while maintaining or reducing costs, equalizing access to care, and increasing visibility as a healthcare system that offers a virtual obstetric experience. The first phase of deployment was in multiple rural locations serving patients that have severe access to care limitations. The next phase of the Banner relationship is currently being scoped and is intended to expand the use cases, locations, and patient populations. |
| ● | Sanford Health. Sanford Health, one of the largest health systems in the United States, is dedicated to the integrated delivery of health care, genomic medicine, senior care and services, global clinics, research, and affordable insurance. Sanford’s healthcare system spans 46 medical centers and over 200 clinics, covering more than 9,000 births annually. Nuvo has completed the first phase of a commercial pilot agreement, that was entered into in January 2023, with Sanford Health that successfully demonstrated the end-to-end operational use of our INVU platform across multiple sites, which included the usability, accuracy, training, logistics and support needs for a patient self-administered model. The next phase of the Sanford relationship is currently being scoped and is intended to expand the use cases, locations, and patient populations across Sanford Health. |
| ● | Sheba Medical Center. We and Sheba Medical Center, or Sheba, in Israel, have a commercial relationship, which was preceded by a Validation Partnership, and prior to that had its long-term goals defined in a non-binding letter of intent on October 31, 2021. The letter of intent contemplated the parties entering into a clinical trial agreements relating to the use of INVU around gestational diabetes management. Sheba is the largest hospital in Israel. The letter of intent contemplated that, together with Sheba’s deployment of INVU, the parties would collaborate on the development of personalized care pathways, predictive/prescriptive analytics, and other care delivery data-management tools, all of which would largely utilize AI in different capacities. The letter of intent additionally contemplated that Sheba sought to eventually adopt INVU within its standard of care protocols, so as to impact the quality of Sheba’s maternal-fetal monitoring and management care. In accordance with the letter of intent, the clinical trial was implemented in 2022, and following that success, a commercial order was placed in December 2022. Our collaboration has recently extended to remote monitoring for extremely high-risk expectant mothers in their homes using the INVU platform amidst the challenges posed by the war between Israel and Hamas. |
PPM Groups and Independent Private Practices – We are also directly targeting large PPM groups and private practices of all sizes. As an example, we have a contract with Nacogdoches Women’s Center (“NWC”), which is administered by one physician with the assistance of registered nurses that provides preventive, diagnostic and therapeutic women’s health and wellness care and comprehensive treatment from adolescence through post-menopause. Many NWC patients live in rural areas and travel long distances to receive medical care, so the ability to provide remote NSTs has been very well-received by NWC providers and patients.
110
Validation Partners
Validation Partners are academic medical institutions that have experience building robust clinical and practical evidence based on our already developed INVU platform to establish evidence of impact across clinical, operational and societal metrics.
| ● | University of Pennsylvania. UPenn has already run two clinical trials for comparative endpoints for MHR, FHR and MUA and has completed an NST operational viability trial seeking to validate our MUA and NST capabilities for home use. See “—Clinical Studies—MHR/FHR Study” and “—MUA Study.” UPenn also applied for and won a National Institutes of Health, or NIH, grant seeking to reduce disparate healthcare outcomes for different racial groups by leveraging our INVU platform, a trial for which has recently been activated. |
| ● | Charite. We entered into an Evaluation Agreement with Charite – Universitatsmedizin Berlin and Berlin Institute of Health at Charite (BIH) (collectively, “Charite”) on September 29, 2021 (the “Charite Agreement”). Charite is one of Europe’s largest university hospitals, affiliated with Humboldt University and Free University Berlin. Under the terms of the Charite Agreement, we have agreed to sponsor a clinical trial within and carried out by Charite in Berlin, Germany (the “Charite Trial”) that focuses on shifting the delivery of care from hospitals to home settings for routine pregnancies, and demonstration of clinical impact such as time saved. In addition, there is a special focus on co-developing a biomarker around predicting, preventing and managing preeclampsia. During the course of the Charite Trial, providers would supply participating patients with INVU units for remote monitoring purposes. Although each party will continue to own its own pre-existing intellectual property that is used during the course of the Charite Trial, any new intellectual property that is jointly developed by the parties would be owned by each party based upon its pro rata development contribution thereto, though the other party will be granted a license thereto under the terms of the Charite Agreement. All preparatory work related to the Charite Trial, including detailed protocol planning, building the Charite team, and budget planning has been completed and we expect the complete protocol package to be submitted in the coming weeks. |
New Care Pathway Partners
New Care Pathway Partners are research experts, mainly academic centers, with specific domain expertise that have the goal of advancing pregnancy care and have the ability to analyze our rich and robust data signals to help determine predictive markers through such data. While Charite, which we consider to be both a Validation Partner and New Care Pathway Partner, the University of Utah, Columbia University, Georgia State and Hadasit are currently our only new care pathway partners, we believe we have strong relationships with other potential partners with whom we are in discussions with to develop other predictive markers for such indications as diabetes, preeclampsia and mood disorders, for example.
| ● | University of Utah. The University of Utah has used our technology to test whether certain HR measurements during a pregnancy were indicative of mood disorder and whether mood disorder could be predicted by appropriate pregnancy monitoring pursuant to a data transfer and use agreement we and the University of Utah entered into in February 2021. Initial results showed a correlation between HR variability and mood disorder in pregnant women. The study sought to understand how prenatal maternal distress was related to children’s health outcomes. Supported by a NIMH grant, the study, titled the Baby Affect and Behavior (“BABY Study”), analyzed MHR and FHR variability as measured by our INVU platform across more than 300 women in their third trimester of pregnancy in relation to emotion dysregulation, which can occur when an individual is under stress or struggles to regulate her emotional responses to support effective behavior. The University of Utah conducted physiological assessments of mood and stress in the home, conducted newborn neurobehavioral exams at birth and followed up with mothers and infants postpartum at seven and 18 months. Study participants spanned the full range of emotion dysregulation. Data collection concluded in early 2023, and full results are expected in 2024. |
| ● | Columbia University. Columbia is leading research using the INVU platform to determine if digital signatures within the ECG and PCG signal can help identify early indicators of pre-term birth pursuant to a framework agreement entered into in March 2022. Overseen by Professor Catherine Monk, a renowned researcher in this space, the study is being managed by the Perinatal Pathways Lab, a research group within Columbia University. Supported by a NIMH grant, the study will include roughly 200 participants who will be studied across 600 sessions that include each participant having three separate data points captured. |
111
| ● | Georgia State. Georgia State is using our technology to test whether certain HR measurements during a pregnancy are indicative of disparities in health outcomes for Black and Hispanic women, pursuant to a data transfer and use agreement we and Georgia State entered into in June 2023. Supported by a NIMH grant, the study, titled the GLOW Study, plans to analyze MHR and FHR variability as measured by our INVU platform across approximately 400-500 women in their third trimester of pregnancy, and the participants will be assessed once a day for one to two weeks. Nuvo is currently working closely with the team at Georgia State to navigate this research terrain and this program started enrolling patients in early 2024. |
Strategic Partnerships
We recognize the value of ecosystem partnerships to help drive existing market adoption, provide complementary capabilities to the INVU platform, and accelerate new market expansion. We actively partner with organizations that can help us provide additional value to our customers in these areas. Our partnership with Philips is an example of a partnership built to accelerate adoption and scale within hospital systems and networks. See “—Expanded Commercial Partnership.” In addition, we entered into a strategic partnership with Ouma in March 2022, a total maternity telehealth services company, to deliver an innovative joint solution that provides clinical excellence in maternity telehealth and FDA-cleared remote fetal surveillance technology. The joint solution is designed to address some of the biggest challenges in pregnancy care today, by enabling equitable access to care, improving preventative care delivery, and reducing high individual and system-level costs. The joint solution is a “one-stop shop” for remote pregnancy care that includes our INVU platform for remote patient monitoring (“RPM”), including FDA-cleared NSTs, as well as Ouma’s 24/7/365 maternity telehealth clinical services available in all fifty US states, including access to maternal-fetal medicine specialists, midwives, perinatal nurse navigators, lactation consultants, and behavioral health specialists with expertise in perinatal mood disorders. We and Ouma are actively pursuing commercial agreements for the joint solution.
Research and Development
As of May 1, 2024, our research and development, or R&D, team consisted of 27 people, 13 of whom are located in Israel and 14 of whom are located in Ukraine, conducting research and product development activities. We focus on developing our key technology and innovations in-house where we benefit from the expertise of our highly qualified R&D team, which allows us to ensure that the key technologies and innovations used in our INVU platform reflect our core values and mission. Since inception, our R&D team has been working on developing and improving all aspects of data collection on our INVU platform and have optimized the device design, performance and usability for its current uses, including over the course of several clinical studies. Our R&D team is also focused on continuing to enhance our hardware and software, validating other measurements, such as mECG and fECG, among others, and developing and improving all aspects of data management, including data gathering, data harmonization and AI-based and other data analysis.
Our R&D team also assists as needed in clinical or other studies being performed by others using our technology or in conjunction with us. In addition, most of our relationships with obstetrician networks and other strategic relationships that we have established or are establishing provide for scaling up in stages and usually involve pilot programs which our R&D team advises, assists or is involved with. For example, our relationship with Philips includes a jointly developed solution integrating Nuvo’s platform into Philips technologies.
112
Manufacturing and Supply
We have established and validated a fully outsourced manufacturing operation to scale up production capacity that allows us to engage in high volume manufacturing as needed. Pursuant to our manufacturing plan, our printed circuit boards (“PCBs”) are manufactured in China and Israel and fabricated in China, acoustic sensors are sourced from Japan, reusable ECG sensors are sourced from China and accessories are sourced from Israel and China. The products are then shipped to Israel where they are assembled into a complete sensor band. Our operations in Israel have not been affected by the war between Israel and Hamas, however, see “Risk Factors — Risks Related to Israeli Law and Our Operations in Israel — Conditions in Israel, including the ongoing war between Israel and Hamas, and other conflicts in the region, may adversely affect our business, our results of operations and our ability to raise additional funds.” While there is risk involved in relying on a single major supplier for certain distinguishing production elements, our wireless sensor band’s off-the-shelf components are provided by suppliers according to their availability and lead time of supply. Accessories are added in the United States and the product is packaged and ready for delivery to the expectant mother. We have also established a fulfillment center where sensor bands are prepared for reuse after thorough cleaning, quality control testing, fixing (if required) and refurbishing. We believe that, on average, a sensor band should be viable for monitoring approximately 12 expectant mothers over a three-year period before needing to be replaced.
We entered into a Framework Product Design and Production Agreement on October 18, 2015 with Orange S.r.l., an Italian company, and Starry Limited, a Hong Kong company, for our manufacturing needs. On August 8, 2018, we entered into a partnership with Flextronics Medical Sales and Marketing, Ltd., of Israel, to further support our manufacturing needs. Flextronics is a global manufacturer with specific medical device expertise. Flextronics purchases most of the sensor belt components, tests them, performs the PCB assembly, executes final assembly of the device and ships it to the United States. We also sourced some of the components in our 2021 production batches directly from third parties based on availability. Starry handles FAB assembly in China. We also entered into a Master Services Agreement with SEKO Worldwide, LLC on February 12, 2021 for our warehousing and other needs, which will continue for an initial term of two years and thereafter on a month-to-month basis. SEKO provides global solutions for medical device logistics, specializing in transportation, logistics, forwarding and warehousing. SEKO’s warehouse that we are planning to utilize is located in Pittsburgh, Pennsylvania. SEKO receives the devices from Flextronics and stocks them until fulfillment to expectant mothers. Final packing is made in accordance with the provider’s prescription for the expectant mother. The wireless sensor bands are sent back to SEKO at the end of the monitoring period.
Our manufacturing process and our manufacturing partners’ manufacturing processes and facilities are designed to comply with the FDA’s Quality Systems Requirements and enable us to market our product. Each of these facilities operates in conformance with a variety of International Organization for Standardization (“ISO”) certifications. Nuvo’s CE submission included three BSI audits which found no major nonconformities.
Sales and Marketing
We have an experienced management team with medical technology, women’s health, medical or healthcare, data science, marketing, financial, consumer products, clinical, regulatory, manufacturing, human resources and commercial expertise. We also have developed a U.S. team, consisting primarily of sales and marketing employees, product specialists, and service staff, as well as employees with operational and medical expertise, to carry out our sales and marketing plans in the United States under the guidance of our experienced management team. To achieve this goal, we are devoting considerable resources to commercializing our INVU platform initially in the United States, before we expand our sales and marketing efforts globally. We also intend to start marketing in Europe shortly after receiving a CE mark, which is not guaranteed and may take longer than expected. We believe that our INVU platform can become a global solution and we ultimately intend to become a leader in remote pregnancy monitoring.
We are still in the early stages of commercializing our INVU platform. As of now, our sales and marketing team consists of our VP of Marketing, who has significant experience in sales and marketing with consumer and medical product companies and our business development team in Israel. We are shifting the center of gravity to the United States for U.S. sales and marketing as indicated below. Current marketing efforts include attending scientific and industry conferences, leveraging our Scientific Advisory Board’s network, limited press releases and public relations, information on our website, social media postings and targeted outreach, currently primarily to specific providers. We plan to develop our team gradually as needed. At this stage, we are focusing initially on sales and marketing in the United States directed at healthcare systems and physician practice management groups that we believe will be most effective at implementing our technology into clinical practice. Our initial approach is for implementers such as obstetrician-physician practice management groups and traditional healthcare systems that see a high volume of HRPs, have affiliate health plans or strength in value-based care. We are also targeting validators, or some of the most prestigious academic medical university systems, to work with us to validate specific use cases that demonstrate the highest impact of improved health outcomes and reduced cost of care. We have signed over a dozen commercial contracts with health systems, large private practice groups and independent women’s health practices in the United States and Israel. Additionally, we will continue our current marketing activities with a view to increase consumer awareness. We believe that increasing consumer interest could have a positive impact on provider acceptance.
113
Over the longer term, we aim to demonstrate through our work with these types of systems and the data we gather that we improve the quality of care and reduce costs. We intend to demonstrate this most convincingly through clinical trials, which we are developing together with our validators, which we believe, if successful, will make our implementers more comfortable to implement and integrate our new care pathways. We will also correspondingly track the results our implementers obtain in expectant mother and clinician satisfaction, compliance, cost and outcomes to establish that we improve the quality of care and reduce costs. Once we can provide impact evidence of improved outcomes and reduced hospitalization and other costs, we believe that this will positively affect the ability of our provider partners to incentivize payers such as insurers and self-insured employers to enter into value contracts with them and to become partners, and will incentivize the payers to encourage their obstetrician networks and expectant mothers to utilize our services. Payers have a vested interest in keeping their insured or employees healthy and reducing costs. Finally, we intend to add data partners and to work with them, our clinicians and payers to gather rich and robust data anytime and anywhere through an AI-enabled platform, obtain useful content, develop, expand and improve our databases and, ultimately identify phenomena and develop screening and predictive models, as well as population health strategies. We believe that the data we obtain and the abilities we currently have and are developing with respect to such data will make us an important player in the maternal and fetal health and pregnancy management world. The thrust of our commercial effort will be business to business, but we intend to also focus on increasing consumer awareness.
Reimbursement and Payment
We are receiving payment directly from providers, including clinicians, hospitals and other healthcare facilities that perform pregnancy tests for our INVU platform and services and we do not directly bill any third-party payers. Rather, care providers bill the applicable CPT code against the procedures run on our INVU platform. For example, our healthcare provider customers bill for each use of the INVU platform (e.g., for each NST under CPT code 59025). Their payers then reimburse the care provider for the procedure. We are considering different types of payment plans, but initially we are utilizing introductory rates to help build a provider base. Our payment terms may vary from customer to customer. In the United States, providers receive payment for care of mothers from third-party payers, including private insurers and government insurance programs.
The baseline metrics for protocols for the standard of care and our offering model were validated by a leading health consulting agency in 2018. The baseline reimbursement rates for relevant codes were validated by Navigant, another leading health consulting agency in 2018. Presently, reimbursement for provider services, including the cost of our services, during a measurement period may be made under global codes to the provider under a prospective payment system that bundles services into groups for the purposes of payment using a number of factors, including, among other things, the principal diagnosis, major tests and procedures, status of the treatment, maternal age and complicating secondary diagnosis. Classifications are used in both acute and chronic care settings and employed by both private insurers and government payers. Under global codes, rather than paying the provider for what it spent caring for an expectant mother and unborn baby, payers pay a fixed amount based on the classification. Reimbursement may also be made under procedural codes, which are billed every time a procedure occurs, such as NSTs.
Some payers have begun specifying coverage for at-home NSTs for their patient populations. For example, Home State Health, a managed Medicaid payer in Missouri, recently issued a statement that was sent to all their healthcare providers in the state confirming that CPT Code 59025 is reimbursable with a place of service modifier (12) for at-home monitoring with remote monitoring solutions that are FDA cleared for on-label use for monitoring FHR, MHR and MUA.
Simultaneously with commercial growth, we are working with leading academic institutions to validate some of the benefits of our INVU platform, which we believe to include improved health outcomes and lower costs. As clinical evidence from the volume of expectant mothers being monitored with our INVU platform develops enough to demonstrate improved health outcomes, reduced cost of care and significant volume, we plan to establish partnerships with payers, either directly or in combination with our provider partners. From this data and evidence, we hope to be able to substantiate the specific average savings a payer can expect when our INVU platform is used in certain types of pregnancies.
We then intend to partner with provider groups that are capable of working with and managing the pregnancy journey or certain portions thereof with our INVU platform to be more cost-effective to approach payers and to negotiate value contracts with incentivized terms to deliver more cost-effective obstetric monitoring and management. Value contracts may take many forms, including without limitation, shared savings or per member per month, although there is no certainty as to how these contracts will be negotiated or the form that they will take. We believe that under this later payment model we would negotiate to receive a portion of the earnings of the providers from value contracts.
114
Privacy/Data Security
Numerous state, federal and foreign laws and regulations govern the collection, dissemination, use, processing access to, confidentiality and security of critical or sensitive personal information. In the United States, numerous federal and state laws and regulations, including state data breach notification laws, federal and state consumer protection laws and regulations (for example, HIPAA, or Section 5 of the Federal Trade Commission Act), which governs the collection, use, disclosure, and protection of personal information could apply to the data we collect from users (i.e., expectant mothers) of our INVU platform, as well as the providers who access such data. In particular, regulations promulgated pursuant to HIPAA establish privacy and security standards that limit the use and disclosure of individually identifiable health information, known as protected health information (“PHI”), and require the implementation of administrative, physical and technological safeguards to protect the privacy of protected health information and ensure the confidentiality, integrity and availability of electronic protected health information. In addition, several states have also promulgated and passed privacy laws over the last couple years, the majority of which include provisions that specifically focus on health data that is both collected and processed by third parties. Determining whether protected health information has been handled in compliance with applicable privacy standards and our contractual obligations can require complex factual and statistical analyses and may be subject to changing interpretation. State laws may be more stringent, broader in scope or offer greater individual rights with respect to PHI than HIPAA, and state laws may differ from each other, which may complicate compliance efforts. Entities that are found to be in violation of HIPAA or one of the state laws as the result of a breach of unsecured PHI/health data, a complaint about privacy practices, or an audit by the U.S. Department of Health and Human Service (“HHS”), or a state’s appointed enforcement arm, may be subject to significant civil, criminal and administrative fines and penalties and/or additional reporting and oversight obligations, as well as significant reputational harm, if required to enter into a resolution agreement and corrective action plan to settle allegations of non-compliance.
European Union (“EU”) member states, Switzerland and other countries have also adopted data protection laws and regulations that impose significant compliance obligations for companies collecting and/or processing personal data of EU residents. For instance, the collection and use of personal health data in the EEA/UK is governed by the provisions of the General Data Protection Regulation (“GDPR”), and the GDPR as transposed into the laws of the UK (“UK GDPR”). The GDPR became effective on May 25, 2018 and imposes strict obligations and restrictions on the ability to process, collect, analyze, and transfer personal data. In particular, these obligations and restrictions concern the consent of the individuals (i.e., the data subjects) to whom the personal data relates, the information provided to the individuals, the transfer of personal data out of the EEA, security breach notifications, security and confidentiality of the personal data, and the imposition of substantial potential fines for breaches of the data protection obligations. Data protection authorities from the different EU member states may interpret the GDPR and national laws differently and impose additional requirements, which add to the complexity of processing personal data in the EU. Compliance with these and any other applicable privacy and data security laws and regulations is a rigorous and time-intensive process, and we may be required to put in place additional mechanisms to ensure compliance with the new data protection rules. See Item 3.D. “Risk Factors—Risks Related to Our Business and Our INVU Platform—Our collection, use, storage, disclosure, transfer and other processing of personal information, could give rise to significant costs, liabilities and other risks, including as a result of investigations, inquiries, litigation, fines, legislative and regulatory action and negative press about our privacy and data protection practices, which may harm our business, financial conditions, results of operations and prospects.”
We have invested significant resources in building a compliance-centric platform that is designed to comply with HIPAA and the GDPR/UK GDPR with strong privacy and data security protocols, including the bifurcation of our system architecture between user customer relationship management (“CRM”) data and diagnostic data, all under the day-to-day auspices of our chief privacy and security officer. Among other important steps undertaken by us, we have (i) obtained ISO 27001 certification, (ii) deployed rigorous encryption and other information security measures for both data at rest and data in transit, (iii) established a comprehensive privacy and data security training program for all employees and (iv) adopted a thorough collection of global data privacy and information security policies, and we are continually monitoring the security and stability of our computing environments and systems.
115
Competition
Overview
The pregnancy monitoring and management industry is highly competitive. Traditionally, most of this activity has taken place at an obstetrician’s office, in a hospital or at another healthcare facility. Since their inception, Doppler ultrasound, for monitoring FHR, and TOCO, for monitoring MUA, have dominated as the standards of care for pregnancy monitoring, and each has its own limitations. The Doppler ultrasound cannot be used frequently and continuously under medical guidelines as it is an active technology sending signals into the womb and only provides high-level shallow data. TOCO has proven to be highly inaccurate, with a failure rate of approximately 35%. These tools are commonly combined as a CTG, which delivers both HR and MUA readings in one session. CTG monitoring in most cases requires the expectant mother to be in a clinical setting and physically linked to the transducer with cables and wires. Though acting as the current standard of care, it usually requires an expert to administer and produces limited insights into fetal and maternal wellbeing. While many healthcare facilities and clinicians also have access to electronic fetal monitoring (“EFM”) devices that utilize ECG technology and are more accurate than CTG, depending on the device, these devices are FDA-cleared for use from the 36th week of pregnancy through the intrapartum period. EFM devices are rarely used for pregnancy monitoring in such facilities due to the limitations of these devices, the cost associated with their operation and the need for more advanced technicians. Today, the standard of care is still Doppler ultrasound. We are not aware of any other monitoring device or biopotential fetal monitoring technology that utilizes PCG to monitor HR, although stethoscopes, mainly digital, high-fidelity stethoscopes, that operate based on acoustic signals are used in rare instances by clinicians for routine tests in developed countries. We have the only device or platform that has been cleared by the FDA for monitoring during the INVU monitoring period that utilizes advanced data modalities, such as ECG and PCG.
In recent years and especially since the outset of COVID-19, remote monitoring has become significantly more important with COVID-19 making it necessary in many situations. Most of the solutions have faced difficulties for various reasons, including ease of use, inability to take multiple measurements, inability to measure as accurately as in a healthcare facility and others. As a result, we compete with both the traditional brick and mortar pregnancy monitoring and management systems, many of which are owned or used by our initial target customer base, providers and several remote monitoring systems.
Currently, the competitive landscape is divided into two distinct directions. First, to capture reliable, accurate, sophisticated, rich and robust data, and second, in line with the movement to distributed care, to distribute access of care via remote use and, in some cases, through a self-administered technology. While some technologies address the data quality issue and some address the distributed care issue, we believe that our INVU platform is the only system that tackles both at once by ensuring a comfortable shift of care to a remote setting while providing the highest-fidelity digital data available through its digital signal processing for obstetric care today. We intend to leverage our distributed care data, along with other data we obtain, to develop sufficient rich and robust data to enhance the results of our analysis of data.
We believe that our advanced technology gives us strong advantages as we compete against other companies and systems, and we intend to compete on that basis. We believe the principal competitive factors in our market are and will include (i) product safety; (ii) expectant mother and clinician experience; (iii) strength of clinical evidence; (iv) economic benefits and cost savings, including reducing the need for hospitalization or medical facilities, neonatal intensive care, certain procedures and other costs; (v) ease-of-use; (vi) reliability and accuracy; (vii) acceptance by treating clinicians and healthcare providers; (viii) effective marketing to and education of expectant mothers, clinicians, healthcare providers, hospitals and ultimately payers; (ix) intellectual property protection; and (x) quality and granularity of data collected and the ability to utilize such data, including the ability to predict certain conditions such as preeclampsia, diabetes, pre-term birth, mood disorders and cardiovascular anomalies, and to develop personalized care protocols and population health strategies.
In addition, we believe our technology is more advanced than existing technologies in many cases. Our INVU platform is a holistic pregnancy care solution with two integrated components, hardware and software. The hardware component comprises a proprietary, wireless sensor band with multi-modality technology, which captures detailed and granular signals that we believe enables our INVU platform, after analysis of the signals, to provide useful data relative to in-office monitoring devices. Our wireless sensor band transmits signals from any location to the software component, a dynamic cloud computing environment that processes and analyzes data and, ultimately, transmits personalized reports on key maternal and fetal health metrics to the expectant mother and her clinician through digital visualization tools.
116
Healthcare Facility Monitoring and Competition
Technology maintained at healthcare facilities include a number of monitoring devices that strive to provide rich and robust data during the intrapartum stage. Primary competitors in the healthcare facility setting include Monica Healthcare, now part of General Electric, Nemo Healthcare and Philips Avalon CL. These systems mostly operate on biopotential signals (although not necessarily ECG or PCG) that provide better quality data, including more accurate and reliable HR data, than Doppler ultrasound, and are more ambulatory in nature than most other existing technology utilized in a healthcare facility. As mentioned above, there are no competitors that utilize PCG to monitor HR, however competitors such as Nemo Healthcare and Monica Healthcare utilize ECG. None of these competitors are able to collect ECG and PCG biopotential signals simultaneously. All three measure each of MHR, FHR and MUA. Some use EHG to measure MUA, instead of TOCO. However, this method, which is a secondary analysis of uterine activity, causes the outputs to appear much different than the current standard of care, which has led to low adoption rates. All have received regulatory approval. However, these must generally be used in a healthcare facility, are only cleared by the FDA for use during the intrapartum period and must be administered by a medical professional. Often more senior medical professionals are required. As a result, these devices are not easily used across the pregnancy journey. However, Nemo has recently gained CE clearance for its technology to be utilized remotely, although other limitations remain such as expensive single-use patches, and cumbersome bases that limit real mobility and portability of use.
The Philips Avalon CL wireless Doppler/TOCO-based technology and fetal monitoring solution also has certain advantages, such as no sensor band, being wireless and a claim that it is able to measure the HR of triplets. It was designed specifically to create a more ambulatory intrapartum experience and does not currently enable home care, and, to our knowledge, Philips is not seeking to enable home care and instead has chosen to establish a strategic partnership with Nuvo to address remote fetal monitoring, as described above.
Remote Systems and Competition
Devices that move in the other direction, seeking to provide distributed care, generally work remotely and are not based on the provision of rich and robust data. Technologies that are “on label” as being remote-use devices, are usually miniaturized CTGs that are repackaged for home use. As these technologies were originally designed and intended to be applied and administered by a medical professional, modification to lay person use is very difficult. For example, a portable Doppler still requires readjustment on the expectant mother’s belly in the event that the unborn baby moves during a monitoring session. These technologies generally cannot gather information from the entire pregnancy or data such as beat by beat HR, or the same quality of contraction measurements and are unable to make similar analyses from the data as we plan to do. Primary competitors in the remote setting include Sense4Baby, Pregnabit, Bloom and Heramed.
Some technologies, such as Sense4Baby, incorporate both transducers of the CTG, including TOCO. TOCO is also highly sensitive to placement, and, as indicated above, has a high error rate. Sense4Baby may be the only device currently cleared by the FDA to offer NSTs, but because of the methodology of measurement, is complicated to use. Pregnabit is similar to Sense4Baby but is much smaller. Heramed’s HeraBeat, which is a sleek, handheld Doppler ultrasound transducer often must be handheld for periods of relatively long duration. In addition, HeraBeat can only measure an average FHR and MHR by Doppler technology in short sessions, but has no-label indication for self-administered NSTs. Bloom, although not currently active, may be able to offer NSTs remotely, but, to our knowledge, is not currently cleared to do so. In order to accomplish this, Bloom is taking the opposite approach by gathering data from third party sources, as opposed to distributed data, and using such data to develop algorithms which will enable it to analyze data obtained from expectant mothers and unborn babies remotely.
117
The Future
We intend to seek clearance to extend our INVU monitoring period as well as report other measurements. If we obtain additional clearances to report other measurements that our INVU platform is able to capture, compute and visualize, we will be able to provide and market additional pregnancy health metrics to participants in the pregnancy care management process, including expectant mothers and clinicians. We intend to utilize the data we collect, combined with external guidelines, to establish cloud-based decision support systems. We intend to develop decision support tools to analyze the data we collect to develop and execute new personalized care protocols and population health strategies which we believe will enhance our value-based care model. We also intend to apply data algorithms and other innovative digital tools to conduct AI-powered machine learning computer analyses to identify patterns and trends based on the data and to develop predictive models to ultimately enable population health strategies.
Competitive Risks
Many of our competitors are large, well-capitalized companies with significantly greater market share, name recognition and resources than we have. They are able to spend more on product development, marketing, sales and other product initiatives than we can and have greater name recognition. These competitors may also have established relationships with clinicians and healthcare providers at our targeted health systems and hospitals and may have existing product approvals from hospital value analysis committees at our targeted customers. In addition to competing for market share, we will also compete against these companies for personnel, including qualified sales and other personnel that are necessary to grow our business. As other companies develop new intellectual property in our market, there is the possibility of a competitor acquiring patents or other rights that may limit our ability to update our technologies and products which may impact demand for our products. See Item 3.D. “Risk Factors—Risks Related to Our Business and Our INVU Platform—Our industry is highly competitive and is subject to technological change, which may result in new products or solutions that are superior to our INVU platform or other future products we may bring to market from time to time. If we are unable to anticipate or keep pace with changes in the marketplace and the direction of technological innovation and customer demands, our technology may become less useful or obsolete and our operating results will suffer.”
Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain patent and other proprietary protection for our commercially important technology, inventions and know-how, including the systems and methods that constitute our INVU maternal/fetal monitoring platform; to defend and enforce our patents; to operate without infringing, misappropriating or violating the proprietary rights of others; and to prevent others from infringing, misappropriating or violating our proprietary rights. We rely on know-how and continuing technological innovation to develop and maintain our competitive position. We also rely on the rights provided by a combination of patent, copyright, trademark and trade secret laws and confidentiality and invention assignment agreements to protect our intellectual property rights. Notwithstanding these efforts, we cannot be sure that patents will be granted with respect to any patent applications we have filed or may license or file in the future, and we cannot be sure that any patents we own or license or patents that may be licensed or granted to us in the future will not be challenged, invalidated, or circumvented, or that such patents will be commercially useful in protecting our technology. For more information regarding the risks related to our intellectual property, please see “Risk Factors—Risks Related to Our Intellectual Property.”
As of May 1, 2024, our innovative technology is protected by an extensive global patent portfolio consisting of 16 issued U.S. utility patents, 10 pending U.S. utility patent applications, 43 issued foreign utility patents, one allowed foreign utility patent application, 13 pending foreign utility patent applications, and one PCT patent application. Our patent portfolio also includes three issued U.S. design patents and seven issued foreign design patents. Our patents cover various aspects of our INVU maternal/fetal monitoring platform:
118
| Type of Patent | Title | Country | Status | Application No. | Grant No. | Exp. Date |
| ECG-Based Fetal Heart Rate Detection (Computer-Implemented Process) | ||||||
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | United States of America | Granted | 14/921,489 | 9,392,952 | 10/23/2035 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | United States of America | Granted | 15/205,620 | 10,111,600 | 10/23/2035 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | China | Granted | 201680012051.3 | ZL 201680012051.3 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Switzerland | Granted | 16761155.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Germany (Federal Republic of) | Granted | 60 2016 061 260.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Denmark | Granted | 16761155.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Spain | Granted | 16761155.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | France | Granted | 16761155.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | United Kingdom | Granted | 16761155.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Netherlands | Granted | 16761155.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | European Patent | Granted | 16761155.7 | 3267884 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Australia | Granted | 2016230825 | 2016230825 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Canada | Granted | 2,979,135 | 2,979,135 | 3/10/2036 |
| Utility | SYSTEMS, APPARATUS AND METHODS FOR SENSING FETAL ACTIVITY | Korea, Republic of (KR) | Granted | 10-2017-7028086 | 1900641 | 3/10/2036 |
| Utility | METHODS AND SYSTEMS FOR TRACKING PHYSIOLOGICAL PARAMETERS OF MOTHER AND FETUS DURING PREGNANCY | United States of America | Application | 18/410,243 | ||
119
| PCG-Based Fetal Heart Rate Detection (Computer-Implemented Process) | ||||||
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | United States of America | Granted | 15/071,915 | 9,642,544 | 10/23/2035 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | United States of America | Granted | 15/439,487 | 10,213,120 | 10/23/2035 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | China | Granted | 201680011312.X | ZL 201680011312.X | 3/15/2036 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | Germany (Federal Republic of) | Granted | 602016029610.1 | 3270775 | 3/16/2036 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | European Patent | Granted | 16764295.8 | 3270775 | 3/16/2036 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | Australia | Granted | 2016231895 | 2016231895 | 3/16/2036 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | Canada | Granted | 2,979,785 | 2979785 | 3/16/2036 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY | Korea, Republic of (KR) | Granted | 10-2017-7029697 | 1834716 | 3/16/2036 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | Germany (Federal Republic of) | Granted | 60 2019 020 066.8 | 3752055 | 2/11/2039 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | France | Granted | 19750926.8 | 3752055 | 2/11/2039 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | United Kingdom | Granted | 19750926.8 | 3752055 | 2/10/2039 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | Netherlands | Granted | 19750926.8 | 3752055 | 2/11/2039 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | European Patent | Granted | 19750926.8 | 3752055 | 2/11/2039 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | United States of America | Granted | 16/969,106 | 11,877,834 | 5/25/2041 |
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | China | Allowed | 201980025158.5 | ||
| Utility | SYSTEMS, APPARATUSES AND METHODS FOR SENSING FETAL ACTIVITY (“Improved PCG Algorithm”) | United States of America | Application | 18/519,123 | ||
120
| ECG-PCG Fetal Heart Rate Fusion (Computer-Implemented Process) | ||||||
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | United States of America | Granted | 15/072,051 | 9,572,504 | 10/23/2035 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | United States of America | Granted | 15/389,618 | 10,039,459 | 10/23/2035 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | China | Granted | 201680011539.4 | ZL 201680011539.4 | 3/15/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | European Patent | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Switzerland | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Germany (Federal Republic of) | Granted | 16782707.0 | 602016035948.0 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Denmark | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | France | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | United Kingdom | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Netherlands | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Norway | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Sweden | Granted | 16782707.0 | 3270774 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Australia | Granted | 2016252353 | 2016252353 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Canada | Granted | 2,979,953 | 2,979,953 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Israel | Granted | 254499 | 254499 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Japan | Granted | 2017-559639 | 6,457,117 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | Korea, Republic of (KR) | Granted | 10-2017-7029603 | 1933338 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | India | Granted | 201747036415 | 491089 | 3/16/2036 |
| Utility | CONTINUOUS NON-INVASIVE MONITORING OF A PREGNANT HUMAN SUBJECT | United States of America | Application | 18/470,007 | ||
121
| ECG-Based Uterine Activity Detection (Computer-Implemented Process) | ||||||
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | United States of America | Granted | 16/529,696 | 10,772,568 | 8/1/2039 |
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | Australia | Granted | AU2019313480 | 2019313480 | 8/1/2039 |
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | Japan | Granted | 2021-505374 | 7116247 | 8/1/2039 |
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | Canada | Granted | 3,108,360 | 3,108,360 | 8/1/2039 |
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | China | Granted | 201980064893.7 | ZL 201980064893.7 | 7/31/2039 |
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | European Patent | Published | 19844562.9 | ||
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | India | Published | 202147008231 | ||
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | Israel | Application | 280538 | ||
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | United States of America | Application | 18/610,724 | ||
| PCG-Based Uterine Activity Detection (Computer-Implemented Process) | ||||||
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | United States of America | Granted | 17/168,771 | 11,284,833 | 2/5/2041 |
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | United States of America | Published | 17/817,622 | ||
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | China | Published | 202180023476.5 | ||
| Utility | SYSTEMS AND METHODS FOR MATERNAL UTERINE ACTIVITY DETECTION | European Patent | Published | 21750658.3 | ||
| ECG-PCG Uterine Activity Fusion (Computer-Implemented Process) | ||||||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | United States of America | Granted | 17/324,947 | 11,324,437 | 2/5/2041 |
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | Australia | Application | 2021217206 | ||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | Canada | Application | 3,170,821 | ||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | China | Published | 202180023452.X | ||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | European Patent | Published | 21750565.0 | ||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | Israel | Application | 295417 | ||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | Japan | Published | 2022-548054 | ||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | Korea, Republic of (KR) | Application | 10-2022-7030546 | ||
| Utility | FUSION SIGNAL PROCESSING FOR MATERNAL UTERINE ACTIVITY DETECTION | United States of America | Published | 18/623,247 | ||
122
| Self-Adjusting Belt (Device and Method) | ||||||
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | United States of America | Granted | 16/279,665 | 10,617,355 | 2/19/2039 |
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | United States of America | Granted | 16/846,836 | 11,534,109 | 2/19/2039 |
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | European Patent | Granted | 19754473.7 | 3755988 | 2/19/2039 |
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | Germany (Federal Republic of) | Granted | 19754473.7 | 3755988 | 2/19/2039 |
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | France | Granted | 19754473.7 | 3755988 | 2/19/2039 |
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | United Kingdom | Granted | 19754473.7 | 3755988 | 2/19/2039 |
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | Netherlands | Granted | 19754473.7 | 3755988 | 2/19/2039 |
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | China | Published | 201980026482.9 | ||
| Utility | SYSTEM AND METHOD FOR MAINTAINING SENSOR CONTACT | United States of America | Application | 18/416,232 | ||
| ECG-Based Prenatal Tracking and Clinical Decision Support (Computer-Implemented Process) | ||||||
| Utility | SYSTEMS, DEVICES, AND METHODS UTILIZING BIO-POTENTIAL DATA OBTAINED BY A PLURALITY OF BIO-POTENTIAL SENSORS FOR PRENATAL TRACKING | United States of America | Granted | 18/161,789 | 11,972,868 | 1/30/2043 |
| Utility | SYSTEM, DEVICE, AND METHOD FOR PRENATAL CLINICAL DECISION SUPPORT | Patent Cooperation Treaty | Published | PCT/IB2023/000034 | 9/28/2024 | |
| Utility | SYSTEMS, DEVICES, AND METHODS UTILIZING BIO-POTENTIAL DATA OBTAINED BY A PLURALITY OF BIO-POTENTIAL SENSORS FOR PRENATAL TRACKING | United States of America | Application | 18/621,219 | ||
| Dry Electrode (Device) | ||||||
| Utility | ELECTRODES FOR ABDOMINAL FETAL ELECTROCARDIOGRAM DETECTION | United States of America | Granted | 15/071,956 | 9,763,583 | 10/23/2035 |
| Acoustic Sensor (Device) | ||||||
| Utility | ACOUSTIC SENSORS FOR ABDOMINAL FETAL CARDIAC ACTIVITY DETECTION | United States of America | Granted | 15/071,884 | 9,713,430 | 10/23/2035 |
| Musical Maternity Belt (System and Related Method) | ||||||
| Utility | MUSICAL MATERNITY BELT | United States of America | Granted | 11/834,085 | 8,396,229 | 8/6/2027 |
| Big Data (Computer-Implemented Process) | ||||||
| Utility | A SPECIALLY PROGRAMMED COMPUTER PLATFORM TO GENERATE NEW DATA RELATED TO A CARRYING MOTHER AND METHODS OF USE THEREOF | United States of America | Application | 63/560,796 | 3/3/2024 | |
| Assessing Amniotic Fluid Volume (Computer-Implemented Process) | ||||||
| Utility | METHODS FOR ASSESSING AMNIOTIC FLUID VOLUME USING BIOIMPEDANCE TOMOGRAPHY | United States of America | Application | 63/610,435 | 12/15/2023 | |
123
| Sensor Belt Design (First Generation) | ||||||
| Design | BELT | United States of America | Granted | 29/545,494 | D793,027 | 8/1/2032 |
| Design | BELT | China | Granted | 201630178263.1 | ZL 201630178263.1 | 5/13/2026 |
| Design | BELT | European Union | Granted | 3123926 | 003123926-0001 | 5/12/2041 |
| Design | BELT | United Kingdom | Granted | 90031239260001 | 90031239260001 | 5/12/2041 |
| Design | BELT | Korea, Republic of (KR) | Granted | 30-2016-0022442 | 30-0883380-000 | 5/12/2036 |
| Sensor Belt Design (Second Generation) | ||||||
| Design | BELT | United States of America | Granted | 29/606,637 | D880,701 | 4/7/2035 |
| Design | BELT | China | Granted | 201730612839.5 | ZL 201730612839.5 | 12/4/2027 |
| Design | BELT | European Union | Granted | 004538395 | 004538395-0001 | 12/6/2042 |
| Design | BELT | United Kingdom | Granted | 90045383950001 | 90045383950001 | 12/6/2042 |
| Musical Maternity Belt Design | ||||||
| Design | MUSICAL MATERNITY BELT | United States of America | Granted | 29/322,927 | D614,377 | 4/27/2024 |
124
The term of individual patents depends on the legal term for patents in the countries in which they are granted. In most countries, including the United States, the patent term is generally 20 years from the earliest claimed filing date of a nonprovisional patent application in the applicable country. Our issued U.S. and foreign patents are anticipated to naturally expire between 2035 and 2043, and our U.S. pending patent applications and pending PCT applications, if issued into patents, are anticipated to naturally expire between 2035 and 2043, excluding any additional patent term adjustment(s) or extension(s), and assuming payment of all applicable maintenance or annuity fees. Once a patent expires, patent protection ends and an invention enters the public domain allowing anyone to commercially exploit the invention without infringing the patent. Our issued U.S. design patents are anticipated to naturally expire between 2032 and 2035. Our European Community Design Registrations are anticipated to naturally expire between 2041 and 2042. Our issued foreign design patents are anticipated to naturally expire between 2026 and 2042. We plan to continuously explore reasonable opportunities to expand our patent portfolio.
Patents may not be issued from any of our pending applications and our issued patents may not be of sufficient scope or strength to provide meaningful protection for our technology. Notwithstanding the scope of the patent protection available to us, a competitor could develop methods or devices that are not covered by our patents or circumvent these patents. Furthermore, numerous U.S. and foreign-issued patents and patent applications owned by third parties exist in the fields in which our maternal/fetal monitoring platform competes. Because patent applications can take many years to publish, there may be applications unknown to us, which may result in issued patents that our existing or future products or technologies may be alleged to infringe.
There has been substantial litigation regarding patent and other intellectual property rights in the medical device industry. We may need to engage in litigation to enforce patents issued to us, to protect our trade secrets or know-how, to defend against claims of infringement of the rights of others or to determine the scope and validity of the proprietary rights of others. Such litigation could be costly and could divert our attention from other functions and responsibilities. Furthermore, even if our patents are found to be valid and infringed, a court may refuse to grant injunctive relief against the infringer and instead grant us monetary damages or ongoing royalties. Such monetary compensation may be insufficient to adequately offset the damage to our business caused by the infringer’s competition in the market. Adverse determinations in litigation could subject us to significant liabilities to third parties, require us to seek licenses from third parties or prevent us from manufacturing, selling or using the product determined to be infringing, any of which could harm our business. See “Risk Factors—Risks Related to Our Intellectual Property” for additional information regarding these and other risks related to our intellectual property portfolio.
We also rely upon trademarks to build and maintain the integrity of our brand. As of May 10, 2023, we owned three registered U.S. trademarks. We also have three trademarks which are registered or granted or for which we have pending trademark applications in the European Union, the United Kingdom, China, Japan and the World Intellectual Property Organization. Further, we own five registered trademarks in Israel. One of the trademarks also has pending applications in Canada and India. The trademarks and trademark applications generally relate to the Company name, INVU and the INVU logo, as well as previous names used in connection with our INVU platform. We also rely, in part, on trade secrets, know-how, continuing technological innovation, and confidential information, to develop and maintain our competitive position and protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. However, such proprietary rights are difficult to protect. We seek to protect our proprietary rights through a variety of methods, including confidentiality and assignment agreements with suppliers, employees, consultants and others who may have access to our proprietary information. However, these agreements may not provide meaningful protection. These agreements may be breached, and we may not have an adequate remedy for any such breach. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have implemented measures to protect and preserve our trade secrets, such measures can be breached, and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors, or misused by any collaborator to whom we disclose such information. Despite any measures taken to protect our intellectual property, unauthorized parties may attempt to copy aspects of our INVU platform or to obtain or use information that we regard as proprietary. As a result, we may be unable to meaningfully protect our trade secrets and proprietary information. For more information regarding the risks related to our intellectual property, please see “Risk Factors—Risks Related to Our Intellectual Property.”
125
Government Regulation
U.S. Food and Drug Administration
We are subject to extensive and ongoing regulation by the FDA under the Federal Food, Drug, and Cosmetic Act of 1938, as amended, and its implementing regulations, or collectively, the FDCA, as well as other federal and state regulatory bodies in the United States. These laws and regulations govern, among other things, product design and development, pre-clinical and clinical testing, manufacturing, packaging, labeling, storage, record keeping and reporting, clearance and approval, marketing, distribution, promotion, import and export, and post-marketing surveillance.
Unless an exemption applies, each new or significantly modified medical device for distribution in the United States will require either a premarket notification to the FDA requesting permission for distribution under section 510(k) of the FDCA or approval from the FDA under the premarket approval, or PMA, process, or grant of a de novo request for classification. Each of the 510(k) premarket notification, de novo and the PMA process can be resource intensive, expensive, and lengthy, as well as require payment of significant user fees, unless an exemption is available.
Device Classification
The FDCA classifies medical devices into one of three classes—Class I, Class II, or Class III—depending on the level of control necessary to assure the safety and effectiveness of the device.
Class I includes devices with the lowest safety risk to the user and are those for which safety and effectiveness can be reasonably assured by adherence to a set of FDA regulations, referred to as the General Controls for Medical Devices, or general controls, which require compliance with the applicable portions of the FDA’s Quality System Regulation (“QSR”), facility registration and product listing, reporting of adverse events, and product problems, and truthful and non-misleading labeling and, in some cases, marketing materials. Although most Class I devices are exempt from the premarket notification process, some Class I or low risk devices also require premarket clearance by the FDA through the 510(k) premarket notification process described below.
Class II devices are those that are subject to general controls and special controls, such as performance standards, post-market surveillance, patient registries, development and dissemination of guidance documents, and recommendations from the FDA. Most Class II devices require premarket clearance by the FDA through the 510(k) premarket notification process described below. Some Class II devices, however, are exempt from the premarket notification process.
Class III devices are devices that require a PMA. Class III devices include devices that are novel and not substantially equivalent to a predicate device, as well as those devices that pose the greatest risk to safety such as life-supporting or life-sustaining devices and implantable devices. Because the safety and effectiveness of Class III devices cannot be assured by general controls and special controls, these devices must undergo the PMA process, which is generally more costly and time-consuming than the 510(k) premarket notification process. A PMA application typically includes, but is not limited to, extensive technical information, non-clinical laboratory studies, labeling, and financial disclosure information for the clinical investigators in the device studies. Additionally, the PMA application must provide valid clinical evidence that demonstrates to the FDA’s satisfaction a reasonable assurance of the safety and effectiveness of the device for its intended use.
The Investigational Device Exemption Process
In the United States, absent certain limited exceptions, human clinical trials intended to support medical device clearance or approval with the FDA require an investigational device exemption (“IDE”) application. An IDE allows the device to be used in a clinical trial to collect safety and effectiveness data. Clinical studies are most frequently conducted to support a PMA. Only a small percentage of 510(k) premarket notifications require clinical data to support the application. All clinical evaluations of medical devices, unless exempt, must have an approved IDE from the FDA before the study is initiated.
126
If the device presents a “significant risk,” as defined by the FDA, to human health, the FDA requires the device sponsor to submit an IDE application to the FDA, which must be approved prior to commencing clinical trials. A significant risk device is one that presents a potential for serious risk to the health, safety or welfare of a patient and either is implanted, purported or represented to be used in supporting or sustaining human life; is for a use that is substantially important in diagnosing, curing, mitigating or treating disease or otherwise preventing impairment of human health; or otherwise presents a potential for serious risk to a subject.
An IDE application requires certain data for support, such as animal and laboratory testing results that show the device is safe for human clinical trials. Generally, clinical trials for a device begins once the FDA approves the IDE application, and an IRB approves the clinical trial’s protocol and informed consent for trial subjects. The IRB is responsible for the initial and continuing review of the IDE, and may pose additional requirements for the conduct of the study.
If the FDA determines that there are deficiencies or other concerns with an IDE for which it requires modification, the FDA may permit a clinical trial to proceed under a conditional approval. Submission of an IDE application does not assure that human clinical trials will be allowed to commence. Furthermore, the FDA’s approval of an IDE application does not bind the FDA to accept the results of a clinical trial as sufficient proof of the device’s safety and effectiveness.
If the device is considered a “nonsignificant risk” device, IDE submission to FDA is not required. Instead, only approval from the IRB overseeing the investigation at each clinical trial site is required. Abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent from subjects, and labeling and record-keeping requirements also apply to non-significant risk device studies.
All clinical trials also must be conducted in accordance with the FDA’s IDE regulations governing investigational device labeling, prohibition of promotion, record keeping, and reporting and monitoring responsibilities of the clinical trials’ sponsors and investigators. Clinical trials must further comply with the FDA’s good clinical practice regulations for IRB approval, informed consent and other human subject protections. If a human clinical trial receives any U.S. government funding or support, such as a grant or cooperative research and development agreement, then the clinical trial must also comply with the HHS Office for Human Research Protections regulations for the protection of human subjects in research, including the Common Rule.
If a clinical trial is commenced, its results may be unfavorable or, even if the intended safety and effectiveness success criteria are achieved, may not be considered sufficient by the FDA to grant marketing approval or clearance. The commencement or completion of any clinical trial may be delayed or halted, or be inadequate to support a clearance or approval for numerous reasons, including, but not limited to, the following:
| ● | The FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold; |
| ● | Patients do not enroll in clinical trials at the rate expected; |
| ● | Patients do not comply with trial protocols; |
| ● | Patient follow-up is not at the rate expected; |
| ● | Patients experience adverse events; |
| ● | Patients die during a clinical trial, even though their death may not be related to the products that are part of the trial; |
| ● | Device malfunctions occur with unexpected frequency or potential adverse consequences; |
| ● | Side effects or device malfunctions of similar products already in the market that change the FDA’s view toward approval of new or similar clearances or approvals or result in the imposition of new requirements or testing; |
127
| ● | IRBs and third-party clinical investigators may delay or reject the trial protocol; |
| ● | Third-party clinical investigators decline to participate in a trial or do not perform a trial on the anticipated schedule or consistent with the clinical trial protocol, investigator agreement, investigational plan, good clinical practices, the IDE regulations or other FDA or IRB requirements; |
| ● | Third-party investigators are disqualified by the FDA; |
| ● | We or third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate manner or consistent with the clinical trial protocol or investigational or statistical plans, or otherwise fail to comply with IDE regulations governing responsibilities, records and reports of sponsors of clinical investigations; |
| ● | Third-party clinical investigators have significant financial interests related to us or our study such that the FDA deems the study results unreliable, or we or third-party clinical investigators fail to disclose such interests; |
| ● | Regulatory inspections of our clinical trials or manufacturing facilities may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials; |
| ● | There are changes in government regulations or administrative actions; |
| ● | The interim or final results of the clinical trial are inconclusive or unfavorable as to safety or effectiveness; or |
| ● | The FDA concludes that our trial design is unreliable or inadequate to demonstrate safety and effectiveness. |
The 510(k) Premarket Notification Pathway
Currently, our products are categorized as Class II devices and subject to the premarket notification requirements under section 510(k) of the FDCA. A 510(k) premarket notification submission requires the submitter to demonstrate that the submitter’s device is “substantially equivalent” to a legally marketed device, which is known as a “predicate device.” A predicate device may include a device that was legally marketed prior to May 28, 1976 (a pre-amendment device), a device that has been reclassified from Class III to Class II or Class I or a device that was found substantially equivalent through the 510(k) process. A device is substantially equivalent if, with respect to the predicate device, it has the same intended use and has either (1) the same technological characteristics or (2) different technological characteristics but the information provided in the 510(k) submission demonstrates that the device does not raise new questions of safety and effectiveness and is at least as safe and effective as the predicate device. A showing of substantial equivalence may in some instances require clinical data. Once the 510(k) submission is accepted for review, the FDA has 90 calendar days to review and issue a determination. However, the FDA review often takes longer. Upon review, the FDA may require additional information, including clinical data.
Before the FDA will accept a 510(k) submission for substantive review, the FDA will first assess whether the submission satisfies the minimum threshold of acceptability. If the FDA determines that the 510(k) submission is incomplete, the FDA will issue a “Refuse to Accept” letter that generally outlines the information the FDA believes is necessary to permit a substantive review and to reach a determination regarding substantial equivalence. A submitter must present the requested information within 180 days before the FDA will proceed with additional review.
Once the FDA determines that the device is substantially equivalent to a predicate device currently on the market, it will send a letter finding substantial equivalency allowing the commercial marketing of the device. If the FDA determines that the device is not substantially equivalent to a predicate device, then the submitter may resubmit another 510(k) with new data, request a Class I or II designation through the FDA’s de novo classification process, file a reclassification petition or submit a PMA application. In the event the FDA determines that the information provided in a 510(k) submission is insufficient to demonstrate substantial equivalence to a predicate device, the FDA generally informs the submitter of the specific information needed for the FDA to make a determination on substantial equivalence. The submitter may then provide the requested information within the time allotted by the FDA or in a new 510(k) submission.
128
After a device receives 510(k) marketing clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change or modification in its intended use, will require a new 510(k) premarket notification submission or, depending on the modification, PMA approval. The determination as to whether or not a modification could significantly affect the device’s safety or effectiveness is initially left to the manufacturer using available FDA guidance. Minor modifications may be submitted to the FDA through a “letter to file” in which the manufacturer documents the rationale for the change and an explanation that a new 510(k) premarket notification submission is not required. The FDA, however, may review such letters to file to evaluate the regulatory status of the modified product at any time and may subsequently require the manufacturer to cease marketing and recall the modified device until the manufacturer submits a new 510(k) premarket notification or obtains a PMA. The FDA may also impose significant regulatory fines or penalties.
Over the years, the FDA has proposed reforms to the 510(k) premarket notification and future proposals could include increased requirements for clinical data and a longer review period, or could make it more difficult for manufacturers to utilize the 510(k) premarket notification process for their products. For example, in November 2018, FDA officials announced forthcoming steps that would modernize the 510(k) premarket notification process. Among other proposals, the FDA announced that it planned to develop proposals to drive manufacturers utilizing the 510(k) premarket notification process toward the use of newer predicate devices, to potentially sunset certain older devices that were used as predicate devices and to potentially publish a list of devices that have been cleared on the basis of demonstrated substantial equivalence to predicate devices that are more than ten years old. These proposals have not yet been finalized or adopted, and the FDA may work with Congress to implement such proposals through legislation. More recently, in September 2019, the FDA published revised final guidance to describe an optional “safety and performance based” premarket review pathway for manufacturers of “certain, well-understood device types” to demonstrate substantial equivalence under the 510(k) clearance pathway, by demonstrating that such device meets objective safety and performance criteria established by the FDA, obviating the need for manufacturers to compare the safety and performance of their medical devices to specific predicate devices in the clearance process. The FDA maintains a list of device types appropriate for the “safety and performance based pathway” and has continued to develop product-specific guidance documents that identify the performance criteria for each such device type, as well as the testing methods for such devices where feasible.
De Novo Classification
If the FDA has not previously classified a medical device as Class I, II or III, then the medical device is automatically classified as Class III regardless of the level of risk it poses. A manufacturer whose novel device is automatically classified as Class III may request classification of its medical device as Class I or Class II through the de novo process on the basis that the device presents low to moderate risk, rather than requiring the submission and approval of a PMA application. A medical device may be eligible for de novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent, or a manufacturer may request de novo classification directly without first submitting a 510(k) premarket notification. The FDA must classify the device within 120 calendar days following receipt of the de novo classification application, although in practice, the FDA’s review may take significantly longer. During the pendency of the FDA’s review, the FDA may issue an additional information letter, which places the de novo classification request on hold and stops the review clock pending receipt of the requested additional information from the manufacturer. In the event the de novo classification requestor does not provide the requested information within 180 calendar days, the FDA will consider the de novo request to be withdrawn. If the manufacturer seeks reclassification as Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject the de novo classification request if it identifies a legally marketed predicate device that would be appropriate for a 510(k) premarket notification or determines that the device is not low to moderate risk or that general controls would be inadequate to control the risks and special controls cannot be developed. In the event the FDA determines the data and information submitted demonstrate that general controls or general and special controls are adequate to provide reasonable assurance of safety and effectiveness, the FDA will grant the de novo classification request and classify the device as either Class I or Class II. Upon device classification, the FDA authorizes the device to be marketed and allows the device to serve as a predicate device for future 510(k) premarket notifications.
129
The PMA Process
Although our products are Class II devices and require premarket notification under section 510(k) of the FDCA, we may in the future be required to undergo the PMA process for one or more products. This process begins with the submission of a PMA application to the FDA. Upon receipt of a PMA application, the FDA will determine whether the application is suitable for filing by reviewing the application for the information required by the PMA regulations and FDA PMA filing policy. If the application does not meet a minimum threshold of acceptability, then the FDA will refuse to file the PMA application. In that event, the FDA will advise the applicant of the information to be provided, or steps needed to be taken, to make the application fileable.
Within 45 days of receipt of a PMA application, the FDA will notify the applicant whether the application has been filed. If filed, the FDA will send the applicant a letter and begin its substantive review. The date the FDA accepted a PMA application for filing is the date the PMA is considered filed. Thereafter, the FDA has 180 days to substantively review the PMA application. However, the FDA can extend the 180-day period to last a significantly longer period of time.
During the substantive review, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting responses. The FDA considers a PMA or PMA supplement to have been voluntarily withdrawn if an applicant fails to respond to an FDA request for information or a major deficiency letter within 180 days. Additionally, the FDA may refer the PMA to an outside panel of experts, or an advisory committee, for review and recommendation. If referred to an advisory committee, then the committee will hold a public meeting to review the PMA. Thereafter, the advisory committee will issue a final report containing its recommendation on the PMA. The FDA has the discretion to accept or reject the advisory committee’s recommendation, as well as ask for more information from the applicant.
Prior to approval, the FDA may inspect the clinical trial sites, as well as inspections of the manufacturing facility and processes. The FDA can delay, limit or deny approval of a PMA application for many reasons, such as the following:
| ● | The device may not be shown safe or effective to the FDA’s satisfaction; |
| ● | The data from pre-clinical studies or clinical trials may be found unreliable or insufficient to support approval; |
| ● | The manufacturing process or facilities may not meet applicable requirements; and |
| ● | Changes in FDA approval policies or adoption of new regulations may require additional data. |
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter, or a letter that contains a number of conditions of approval. Upon fulfilling the conditions of approval, the FDA will issue a letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the letter. If the FDA’s evaluation of a PMA application or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. Additionally, the FDA may determine that additional tests or clinical trials are needed, in which case the PMA may be delayed for a period of time while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when the data are available. The PMA process can be expensive, uncertain and lengthy, and a number of devices for which the FDA approval has been sought by other companies have never been approved by the FDA for marketing.
The FDA will require new PMA applications or supplements for modifications that affect the safety and effectiveness of the device that has been approved through the PMA process, including changes to the manufacturing process, equipment or facility, quality control procedures, sterilization, packaging, expiration date, labeling, device specifications, ingredients, materials or design of a device. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the approved PMA application and may or may not require as extensive technical or clinical data or the convening of an advisory panel, depending on the nature of the proposed change.
As a condition of approving a PMA application, the FDA may require a post-approval study, or post-market surveillance, whereby the applicant conducts a follow-up study or follows certain patient groups for a number of years and makes periodic reports to the FDA on the clinical status of those patients when necessary to protect the public health or to provide additional or longer term safety and effectiveness data for the device. The FDA may also require post-market surveillance for certain devices cleared under a 510(k) premarket notification, such as implants or life-supporting or life-sustaining devices used outside a device user facility. The FDA may also approve a PMA application with other post-approval conditions intended to ensure the safety and effectiveness of the device, such as, among other things, restrictions on labeling, promotion, sale, distribution and use.
130
Post-Market Regulation
After the FDA has cleared or approved a device for marketing, numerous and pervasive regulatory requirements continue to apply. These include the following:
| ● | Establishment registration and device listing with the FDA; |
| ● | QSR requirements, which require manufacturers and contract manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the design and manufacturing process; |
| ● | Labeling regulations and FDA prohibitions against the promotion of investigational products, or “off-label” uses of cleared or approved products; |
| ● | Requirements related to promotional activities; |
| ● | Clearance or approval of product modifications to devices marketed under a 510(k) premarket notification, de novo classification or PMA approval that could significantly affect safety or effectiveness or that would constitute a major change in intended use of one of our cleared devices; |
| ● | Medical device reporting requirements, which require that a manufacturer report to the FDA if a marketed device may have caused or contributed to a death or serious injury, or has malfunctioned and the device or a similar marketed device would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur; |
| ● | Correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field corrections, product removals, or recalls if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; |
| ● | The FDA’s recall authority, whereby it can order device manufacturers to recall from the market a product that is in violation of governing laws and regulations; and |
| ● | Post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
Advertising and promotion of medical devices, in addition to being regulated by the FDA, are also regulated by the Federal Trade Commission and by state regulatory and enforcement authorities. Recently, promotional activities for FDA-regulated products have been the subject of enforcement actions brought under healthcare reimbursement laws and consumer protection statutes. Competitors and others can also initiate litigation relating to advertising claims under the federal Lanham Act and similar state laws. In general, if the FDA determines that our promotional materials, which may include our product training, constitutes promotion of an unapproved or uncleared use, then it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved or uncleared use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
The manufacturing process for medical devices is governed by the FDA’s QSR, which covers the methods, facilities and controls for the design, manufacture, testing, production, quality assurance, labeling, packaging, distribution, installation, and servicing of finished medical devices intended for human use. The QSR also requires, among other things, maintenance of a device master file, design history file, device history records, complaint files and adverse incident files. As a manufacturer, we are subject to periodic scheduled and unscheduled inspections by the FDA. Failure to maintain compliance with the QSR requirements could result in the shut-down of, or restrictions on, manufacturing operations and the recall or seizure of products, which would harm our business. The discovery of previously unknown problems with any of our devices, including unanticipated adverse events or adverse events of increasing severity or frequency, whether resulting from the use of the device within the scope of its clearance or off-label by a physician in the practice of medicine, could result in restrictions on the device, including the removal of the product from the market or voluntary or mandatory device recalls.
131
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that we failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, which may result in any of the following sanctions:
| ● | Untitled letters, warning letters, fines, injunctions, consent decrees, and civil penalties; |
| ● | Unanticipated expenditures to address or defend such actions; |
| ● | Customer notifications for repair, replacement or refunds; |
| ● | Recall, withdrawal, administrative detention or seizure of our devices; |
| ● | Operating restrictions or partial suspension or total shutdown of production; |
| ● | Refusal of or delay in granting any of our submissions for 510(k) premarket notification or requests for PMA approval of new devices or modified devices; |
| ● | Operating restrictions, partial suspension or total shutdown of production; |
| ● | Withdrawing 510(k) premarket notification or PMA approvals that are already granted; |
| ● | Refusal to grant export approval for our devices; or |
| ● | Criminal prosecution. |
Emergency Use Authorization
In emergency situations, such as a nationally declared public health emergency, the FDA has the authority to allow unapproved medical products or unapproved uses of cleared or approved medical products to be used in an emergency to diagnose, treat or prevent serious or life-threatening diseases or conditions caused by chemical, biological, radiological or nuclear warfare threat agents when there are no adequate, approved and available alternatives. In such event, any of our products may be authorized for temporary emergency use.
Under this authority, the FDA may issue an emergency use authorization (“EUA”) for an unapproved device if the following four statutory criteria have been met: (1) a serious or life-threatening condition exists; (2) evidence of effectiveness of the device exists; (3) a risk-benefit analysis shows that the benefits of the product outweigh the risks; and (4) no other alternatives exist for diagnosing, preventing or treating the disease or condition. Evidence of effectiveness includes medical devices that “may be effective” to prevent, diagnose or treat the disease or condition identified in a declaration of emergency issued by the Secretary of HHS. The “may be effective” standard for EUAs requires a lower level of evidence than the “effectiveness” standard that FDA uses for product clearances or approvals in non-emergency situations. The FDA assesses the potential effectiveness of a possible EUA product on a case-by-case basis using a risk-benefit analysis. In determining whether the known and potential benefits of the product outweigh the known and potential risks, the FDA examines the totality of the scientific evidence to make an overall risk-benefit determination. Such evidence, which could arise from a variety of sources, may include (but is not limited to) results of domestic and foreign clinical trials, in vivo efficacy data from animal models and in vitro data, as well as the quality and quantity of the available evidence.
Once granted, an EUA will remain in effect until the earlier of (1) the determination by the Secretary of HHS that the public health emergency has ceased or (2) a change in the approval status of the product such that the authorized use(s) of the product are no longer unapproved. After the EUA is no longer valid, the product is no longer considered to be legally marketed and one of the FDA’s nonemergency premarket pathways would be necessary to resume or continue distribution of the subject product.
The FDA also may revise or revoke an EUA if the circumstances justifying its issuance no longer exist, the criteria for its issuance are no longer met or other circumstances make a revision or revocation appropriate to protect the public health or safety.
132
European Union
The European Union regulates medical devices pursuant to the European Union Regulation 2017/745 (“MDR”), which sets forth the basic regulatory framework for medical devices in the European Union. Conformity with the MDR is represented by the CE Mark, which is obtained by meeting minimum standards of performance, safety and quality, and then, according to a device’s classification, compliance with one or more of a selection of conformity assessment routes. When a European Union member state issues a CE Mark, then the device can be sold throughout the entire European Union without further conformance tests. The CE Mark is contingent upon continued compliance with the applicable regulations and quality system requirements.
Federal and State Privacy and Security Laws
HIPAA requires us to comply with standards for the exchange of health information within our Company and with third parties, such as payers, business associates and patients. These include standards for common healthcare transactions, such as claims information, plan eligibility, payment information and the use of electronic signatures; unique identifiers for providers, employers, health plans and individuals; and security, privacy, breach notification and enforcement. Under HIPAA, a “covered entity” includes healthcare providers, healthcare clearing houses and health plans, and a “business associate” is a person or entity, other than a member of the workforce of a covered entity, who performs functions or activities on behalf of, or provides certain services to, a covered entity that involve access by the business associate to protected health information. We are business associates under HIPAA.
HIPAA transaction regulations establish form, format and data content requirements for most electronic healthcare transactions, such as healthcare claims that are submitted electronically. The HIPAA privacy regulations establish comprehensive requirements relating to the use and disclosure of PHI. The HIPAA security regulations establish minimum standards for the protection of PHI that is stored or transmitted electronically. The HIPAA breach notification regulations establish the applicable requirements for notifying individuals, HHS, and the media in the event of a data breach affecting PHI. Violations of the privacy, security and breach notification regulations are punishable by civil and criminal penalties.
The American Recovery and Economic Reinvestment Act of 2009 (“ARRA”) increased the amount of civil monetary penalties that can be imposed for violations of HIPAA, and the amounts are updated annually for inflation. The current penalties for HIPAA violations can range from $127 to $1.919 million per violation, with a maximum fine of $1.919 million for identical violations during a calendar year. ARRA also authorized state attorneys general to bring civil enforcement actions under HIPAA, and attorneys general are actively engaged in enforcement. These penalties could be in addition to other penalties assessed by a state for a breach that would be considered reportable under the state’s data breach notification laws.
The HITECH Act was enacted in conjunction with ARRA. Among other things, the HITECH Act makes business associates of covered entities directly liable for compliance with certain HIPAA requirements, strengthens the limitations on the use and disclosure of PHI without individual authorizations and adopts the additional HITECH Act enhancements, including enforcement of noncompliance with HIPAA due to willful neglect. The changes to HIPAA enacted as part of ARRA reflect a Congressional intent that HIPAA’s privacy and security provisions be more strictly enforced. These changes have stimulated increased enforcement activity and enhanced the potential that healthcare providers and their business associates will be subject to financial penalties for violations of HIPAA. In addition, the Secretary of HHS is required to perform periodic audits to ensure covered entities and their business associates comply with the applicable HIPAA requirements, increasing the likelihood that a HIPAA violation will result in an enforcement action.
In addition to the federal HIPAA regulations, most states also have laws that protect the confidentiality of health information and other personal data. Certain of these laws grant individual rights with respect to their information, and we may be required to expend significant resources to comply with these laws. Furthermore, all 50 states and the District of Columbia have adopted data breach notification laws that impose, in varying degrees, an obligation to notify affected persons and/or state regulators in the event of a data breach or compromise, including when their personal information has or may have been accessed by an unauthorized person. Some state breach notification laws may also impose physical and electronic security requirements regarding the safeguarding of personal information, such as social security numbers and bank and credit card account numbers. Violation of state privacy, security, and breach notification laws can trigger significant monetary penalties. In addition, certain states’ privacy, security, and data breach laws, including, for example, the California Consumer Privacy Act, include a private right of action that may expose us to private litigation regarding our privacy practices and significant damages awards or settlements in civil litigation.
133
Even when HIPAA does not apply, according to the FTC, failing to take appropriate steps to keep consumers’ personal information secure may constitute unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA security regulations.
U.S. Federal, State, and Foreign Fraud and Abuse Laws
The federal and state governments have enacted, and actively enforce, a number of laws to address fraud and abuse in federal healthcare programs, including any healthcare plans or programs that are funded by the United States government (other than certain federal employee health insurance benefits/programs) and certain state healthcare programs that receive federal funds, such as Medicaid. Our business is subject to compliance with these laws.
Federal and State Anti-Fraud and Anti-Kickback Laws
Our operations are subject to various federal anti-fraud and abuse laws, including without limitation the federal Anti-Kickback Statute. The federal Anti-Kickback Statute is a broad criminal statute that, among other things, prohibits the knowing and willful offer, solicitation, receipt, or payment of any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, for the purpose of inducing or rewarding the order, purchase, use or recommendation of items or services that may be paid for, or reimbursed by, in whole or in part, a federal healthcare program, such as Medicare or Medicaid. Further, the term “remuneration” has been broadly interpreted to include anything of value, including cash, improper discounts and free or reduced price items and services. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Almost any financial interaction with a healthcare provider, patient or customer may implicate the federal Anti-Kickback Statute.
Several courts have interpreted the federal Anti-Kickback Statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal health care covered business, the statute has been violated. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Government officials have focused recent federal Anti-Kickback Statute enforcement efforts on, among other things, the sales and marketing activities of medical device manufacturers and other healthcare companies, and recently have brought cases against individuals or entities who allegedly offered unlawful inducements to potential or existing customers in an attempt to procure their business. Judgments and settlements of these cases by healthcare companies have involved significant fines and, in some instances, criminal pleas and convictions. Conviction under the federal Anti-Kickback Statute results in mandatory exclusion from participation in the federal healthcare programs, meaning an entity cannot receive reimbursement from federal healthcare programs or contract with anyone who receives reimbursement from federal healthcare programs. Violators may be subject to, among other things, imprisonment and significant criminal fines up to $100,000 for each violation under the Anti-Kickback Statute, as well as civil monetary penalties up to $100,000 for each violation, plus up to three times the remuneration involved. Further, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute also constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Due to the breadth of these laws, the narrowness of statutory exceptions and regulatory safe harbors available, and the range of interpretations to which they are subject, it is possible that some of our current or future practices might be challenged under one or more of these laws.
Another key federal healthcare law is the federal healthcare fraud statute, which was added by HIPAA. HIPAA created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The HIPAA healthcare fraud statute prohibits, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment and/or exclusion from government-sponsored programs. The HIPAA false statements statute prohibits, among other things, knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony, which requires exclusion from participation in federal healthcare programs, and may result in substantial fines and/or imprisonment. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statutes or specific intent to violate them in order to have committed a violation.
134
Federal law also includes a provision commonly known as the “Stark Law,” which generally (i) prohibits physicians from making referrals for designated health services to entities in which the physicians have a direct or indirect financial relationship, and (ii) prohibits entities from presenting or causing to be presented claims or bills to any individual, third party payer, or other entity for designated health services furnished pursuant to a prohibited referral, unless permitted under a statutory or regulatory exception. Violations of the Stark Law may result in significant civil sanctions, including civil monetary penalties, denial of payment, refunds of amounts collected in violation of the Stark Law and exclusion from Medicare programs.
In addition to these federal laws, there are often similar state anti-kickback and false claims laws that typically apply to arrangements involving reimbursement by a state-funded Medicaid or other healthcare program. Other laws prohibit certain direct or indirect payments or fee-splitting arrangements between healthcare providers and other persons and entities where they are designed to obtain or induce the referral of patients from a particular person or provider. Often, these laws closely follow the language of their federal law counterparts, although they do not always have the same exceptions or safe harbors. In some states, these anti-kickback laws apply with respect to all payors, including commercial health insurance companies. In addition, many states, including California, also have state anti-“self-referral” and other laws that are not limited to Medicare and Medicaid referrals, with which we must comply.
We monitor all aspects of our business and have developed a comprehensive ethics and compliance program that is designed to monitor and address prevention of anti-fraud and kickback laws.
The False Claims Act
The federal False Claims Act (“FCA”) prohibits false claims or requests for payment, for which payment may be made by a federal government program, including healthcare services. Under the FCA, the federal government may penalize any person who knowingly submits, or participates in submitting, claims for payment to the federal government that are false or fraudulent, or which contain false information. Any person who knowingly makes or uses a false record or statement to avoid paying the federal government, or knowingly conceals or avoids an obligation to pay money to the federal government, may also be subject to fines under the FCA. Under the FCA, the term “person” means an individual, company or corporation.
The federal government has used the FCA in connection with Medicare, Medicaid and other governmental program fraud in areas such as violations of the federal Anti-Kickback Statute or the Stark Law, coding errors, billing for services not provided and submitting false cost reports. The FCA has also been used to bring suit against people or entities that bill services at a higher reimbursement rate than is allowed and that bill for care that is not medically necessary. In addition to government enforcement, the FCA authorizes private citizens to bring qui tam or “whistleblower” lawsuits, greatly extending the number of actions under the FCA. As of 2023, the per-claim penalty range was between $13,508 and $27,018.
The Fraud Enforcement and Recovery Act of 2009 (“FERA”) amended the FCA with the intent of enhancing the powers of government enforcement authorities and whistleblowers to bring FCA cases. In particular, FERA attempts to clarify that liability may be established not only for false claims submitted directly to the government but also for claims submitted to government contractors and grantees. FERA also sought to clarify that liability exists for attempts to avoid repayment of overpayments, including improper retention of federal funds. Furthermore, FERA included amendments to the FCA procedures, expanding the government’s ability to use the Civil Investigative Demand process to investigate potential defendants, and permitting government complaints in intervention to relate back to the filing of the whistleblower’s original complaint. FERA has increased both the volume and liability exposure of FCA cases brought against healthcare providers and suppliers.
In the Patient Protection and Affordable Care Act and the Healthcare Education and Reconciliation Act, or, collectively, the ACA, Congress enacted requirements related to identifying and returning overpayments made under Medicare and Medicaid. The Centers for Medicare and Medicaid Services (“CMS”) finalized regulations regarding the so-called “60-day rule,” which requires providers and suppliers to report and return Medicare and Medicaid overpayments within 60 days of identifying the same. A provider or supplier who retains identified overpayments beyond 60 days may be liable under the FCA. “Identification” occurs when a person “has, or should have through the exercise of reasonable diligence,” identified and quantified the amount of an overpayment. The final rule also established a six-year lookback period, meaning overpayments must be reported and returned if a person identifies the overpayment within six years of the date the overpayment was received. Providers and suppliers must report and return overpayments even if they did not cause the overpayment. In addition to the FCA, the federal government may use several criminal statutes to prosecute the submission of false or fraudulent claims for payment to the federal government. Many states have similar false claims statutes that impose liability for the types of acts prohibited by the FCA. As part of the Deficit Reduction Act of 2005 (“DRA”), Congress provided states an incentive to adopt state false claims acts consistent with the federal FCA. Additionally, the DRA required providers who receive $5 million or more annually from Medicaid to include information on federal and state FCAs, whistleblower protections and the providers’ or suppliers’ own policies on detecting and preventing fraud in their written employee policies.
135
Civil Monetary Penalties
The Civil Monetary Penalty Act of 1981 imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal healthcare program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent, or offering or transferring remuneration to a federal healthcare beneficiary that a person knows or should know is likely to influence the beneficiary’s decision to order or receive items or services reimbursable by the government from a particular provider or supplier. The government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the Civil Monetary Penalty Act.
Open Payments
The federal Physician Payments Sunshine Act created the Open Payments Program requiring certain manufacturers of drugs, medical devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually to CMS information related to payments and other “transfers of value” to physicians and teaching hospitals. Additionally, applicable manufacturers must report annually ownership and investment interests held by physicians and their immediate family members. Beginning in 2021, applicable manufacturers will also be required to report information and transfers of value provided to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists and certified nurse-midwives. Failure to submit timely, accurate and complete reports may result in substantial monetary penalties. We are subject to the Open Payments Program, and the information we disclose may lead to greater scrutiny, which may result in modifications to established practices and additional costs. Furthermore, similar reporting requirements have also been enacted in several states. In particular, a number of states have enacted laws that require medical device companies to monitor and report payments, gifts and other remuneration made to physicians and other healthcare providers, and, in some states, marketing expenditures. In addition, some state statutes impose outright bans on certain manufacturer gifts to physicians or other healthcare professionals. Some of these laws, referred to as “aggregate spend” or “gift” laws, carry substantial fines if they are violated. An increasing number of countries worldwide also are adopting, or considering, similar laws.
Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act (“FCPA”) prohibits any United States individual or entity from paying, offering or authorizing payment or offering anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign official, political party or candidate, in order to assist the individual or entity in obtaining or retaining business. The FCPA also obligates publicly traded companies, whose securities are listed in the United States, to comply with accounting provisions that require the maintaining of books and records that accurately and fairly reflect all transactions of the company, including all subsidiaries, international and domestic, if any, and to devise and maintain an adequate system of internal accounting controls for international operations. We face significant risks if we, which includes our employees, contractors, business partners, intermediaries or agents, fail to comply with the FCPA and other laws that prohibit improper payments, offers or promises of payment to foreign governments and their officials and political parties by us and other business entities for the purpose of obtaining or retaining business or other advantages. Any violation of the FCPA and related policies could result in severe criminal or civil sanctions, which could adversely affect our business, results of operations and financial condition.
International Laws
Various European countries have adopted anti-bribery laws for individuals and entities who engage in bribery with public officials that may lead to significant fines and criminal penalties. Violations of these anti-bribery laws, or allegations of any violation, could have a negative impact on our business, operations and reputation. For example, the United Kingdom enacted the Bribery Act of 2010 to combat bribery of British and foreign public officials. Under the Bribery Act of 2010, a bribery occurs when a person offers, gives or promises to give a financial or other advantage to induce or reward another individual to improperly perform certain functions or activities, including any function of a public nature. Penalties under the Bribery Act of 2010 can include imprisonment for up to ten years and substantial fines.
In addition to anti-bribery laws, many nations have enacted privacy laws that impose restrictions on the collection, use, storage, disclosure, transfer and other processing of personal information, including health information. For instance, the European Union adopted the GDPR, which imposes stringent data protection requirements, including more robust disclosures to individuals, a strengthened individual data rights regime, shorted timelines for data breach notifications, limitations on retention of information, increased requirements pertaining to special data categories such as health data, and additional obligations regarding third-party processors in connection with the processing of the personal data. The GDPR also imposes strict rules on the transfer of personal data out of the European Union to the United States and other third-party countries. The GDPR authorizes fines for certain violations of up to 4% of global annual revenue or €20 million, whichever is greater. Additionally, the GDPR provides that European Union Member states may make their own stricter laws and regulations limiting the processing of personal data, including genetic, biometric or health data. In 2018, the United Kingdom enacted the Data Protection Act of 2018 as part of its withdrawal from the European Union to apply the GDPR’s standards to the United Kingdom. All of these laws impact our business. Our failure to comply with these privacy laws or significant changes in the laws restricting our ability to obtain required patient information could significantly impact our business and our future business plans.
136
The United States Medicare and Medicaid Programs
We must comply with regulations promulgated by HHS and CMS that pertain to the Medicare and Medicaid programs. Title XVIII of the Social Security Act establishes the Medicare program to pay for the costs of certain healthcare services and items for eligible individuals. Eligibility for Medicare is based on age, disability or affliction with certain diseases. CMS has established guidelines for the Medicare coverage and reimbursement of certain items and services. Generally, to be reimbursed by Medicare, a healthcare item or service furnished to a Medicare beneficiary must be reasonable and necessary for the diagnosis or treatment of an illness or injury, or to improve the functioning of a malformed body part. The methodology for determining coverage status and the amount of Medicare reimbursement varies based upon, among other factors, the setting in which a Medicare beneficiary received healthcare items and services. Any changes in federal legislation, regulations and policy affecting CMS coverage and reimbursement relative to any procedure using our products could have a material effect.
Title XIX of the Social Security Act establishes the Medicaid program, which is a system of medical assistance for families with dependent children and for aged, blind and disabled individuals who are below certain income thresholds. Though federally created, the Medicaid program is a joint federal-state program. CMS administers the federal portion of the Medicaid program with states establishing additional coverage regulations. Changes to the availability of coverage, method or level of reimbursement for relevant services using our products may have a material effect on us.
The Medicare and Medicaid programs are subject to statutory and regulatory changes, retroactive and prospective rate adjustments, administrative rulings, interpretations of policy, intermediary determinations and government funding restrictions, all of which may materially increase or decrease the rate of program payments to healthcare facilities and other healthcare providers and suppliers.
United States Health Reform
There are continuing efforts to reform governmental healthcare programs by federal and state governments that could result in major changes to healthcare delivery and reimbursement systems on a national and state level. The ACA and other laws and regulations that limit or restrict healthcare reimbursement could adversely impact our customers, resulting in their inability to pay us, or pay us in a timely manner, for our products. Since its passage, there have been numerous challenges in federal courts regarding the constitutionality of the ACA. Most recently, the U.S. Supreme Court held that state and individual plaintiffs did not have standing to challenge the ACA’s minimum essential coverage provision. In so holding, the Supreme Court did not consider the larger constitutional question about the validity of this provision, post-repeal of its associated tax penalty, or the validity of the ACA in its entirety. The ultimate impact of this decision and other efforts to repeal, substantially amend, eliminate or reduce funding for the ACA is unknown. The effect of any major modification or repeal of the ACA on our business, operations or financial condition cannot be predicted, but could be materially adverse.
The ACA has changed healthcare financing and delivery by both governmental and commercial third-party payers and affected medical device manufacturers. The ACA provided incentives to programs that increase the federal government’s comparative effectiveness research, and implemented payment system reforms, including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models.
Since the ACA, other legislative changes have been proposed and adopted. For example, the Budget Control Act of 2011, among other things, included reductions to CMS payments to providers of 2% per fiscal year, which went into effect on April 1, 2013, and, due to subsequent legislative amendments, will remain in effect through 2030. These provisions were suspended from May 1, 2020 through July 1, 2022. Additionally, the American Taxpayer Relief Act of 2012, among other things, reduced CMS payments to several types of providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
We believe that there will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payers to reduce costs while expanding individual healthcare benefits. Some of these changes could impose additional limitations on rates we will be able to charge for our current and future products or the amounts of reimbursement available for our current and future products from governmental agencies or other third-party payers. Current and future healthcare reform legislation and policies could harm our business and financial condition.
137
| C. | Organizational Structure |
Nuvo Group Ltd., a limited liability company organized under the laws of the State of Israel is our wholly-owned subsidiary and Nuvo Group USA, Inc., a Delaware corporation is a wholly-owned subsidiary of Nuvo.
| D. | Property, Plants and Equipment |
We sub-lease approximately 350 square meters for our corporate headquarters located in Tel Aviv, Israel under a rent agreement that expires in December 2024. Pursuant to the agreement, we have an option to terminate the agreement with three months’ notice as of March 30, 2024. We believe that this facility is adequate to meet our current needs in Israel in the near term and that additional space can be obtained on commercially reasonable terms as needed. While our presence in the United States is currently virtual, we may seek office space in the future.
| ITEM 4A. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 5. | OPERATING AND FINANCIAL REVIEW AND PROSPECTS |
Overview
Following and as a result of the Business Combination, the business of Holdco is conducted through Nuvo, its direct, wholly-owned subsidiary.
We believe Nuvo has the potential to become a leader in remote fetal monitoring for pregnancy care. We are leading the transformation from a world where pregnancy care is limited by outdated technology and barriers to accessing care to a world where data-driven, clinically relevant, actionable insights can be accessed both at home and in the clinic, during the INVU monitoring period, by an expectant mother and her clinician. Current poor fetal and maternal health outcomes, limited accessibility to care, and soaring costs all indicate the need for a change in the way that pregnancies are monitored and managed, and we believe Nuvo’s innovative solution, which we refer to as our INVU platform, is the only solution that is positioned to address complete accessibility to care while looking significantly deeper into the pregnancy than standard of care solutions do today. Recognizing that the tools used today to monitor and manage pregnancies may not be the same tools used a decade from now, Nuvo believes its solution is well positioned to be at the forefront of this market shift. Strategically, Nuvo’s platform is currently being commercialized by tapping into a key part of the pregnancy journey, fetal non-stress tests (“NSTs”), by enabling these tests to be conducted remotely with clinical accuracy that has been demonstrated to be equivalent to the standard of care based off of our clinical studies and consumer-grade ease of use. NSTs are medically necessary pregnancy screening procedures that measure fetal heart rate and reaction to movement to assess fetal well-being. NSTs are most commonly conducted with cardiotocography (“CTG”) machines, which were designed for intrapartum monitoring in clinics by experienced healthcare professionals. Through a combination of advanced wearable technology, AI & machine learning, and compelling user experiences (for expectant mothers and clinicians), INVU by NuvoTM (“INVU”) enables increased access to care, deeper insights into maternal-fetal health, reduced clinical staff burden, and improved patient satisfaction.
INVU is composed of a hardware component (wearable), with digital signal processing and cloud analytics, and interfaces for every participant involved in the pregnancy care. The hardware component of our INVU platform is a proprietary self-administered wireless sensor band that clinicians prescribe to expectant mothers who wear the sensor band during virtual visits to capture real-time data on key maternal and fetal health metrics. During these visits, a live reading allows the expectant mother to access simplified data and insights via the paired INVU application. Our wireless sensor band captures a unique set of in-depth physiological data from the expectant mother and unborn baby in a passive manner, without sending energy signals into the womb. Next, the data is digitized and sent wirelessly for analysis on our cloud-based servers by our sophisticated algorithms. Today, when obstetrics clinicians connect to our INVU platform, they have access to a digital dashboard that contains fetal and maternal heart rate and uterine activity tracings recorded during the session and data derived from these measurements for all expectant mothers and unborn babies in their care that use our INVU platform. This data is comparable to the fetal surveillance procedures that normally occur once or twice weekly in the last trimester of pregnancies that have some indication for risk. According to a study in the American Journal of Obstetrics and Gynecology (“AJOG”) analyzing approximately ten million pregnancies, 38% were identified as low risk and 62% were identified as high risk for unexpected complications.
138
The following discussion and analysis summarizes the significant factors affecting the operating results, financial condition, liquidity and cash flows of Nuvo as of and for the periods presented below. The following discussion and analysis should be read in conjunction with Nuvo’s audited financial statements as of and for the years ended December 31, 2023, 2022 and 2021 and the related notes thereto included elsewhere in this Annual Report. Unless otherwise noted, all references in this Item 5 to “we,” “us” or “our” refer to the business of Nuvo prior to the consummation of the Business Combination.
The following discussion and analysis contains forward-looking statements that involve risk and uncertainties, such as statements of our plans, objectives, expectations, and intentions. Our actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under Item 3.D. “Risk Factors” contained in this Annual Report.
| A. | Operating Results |
Components of Our Results of Operations
Revenues
During the year ended December 31, 2023, we started to generate revenues from product sales and services. While still minimal as of that date, we have also entered into a series of commercial contracts which we believe will generate increasing levels of revenue for our business in the future. For additional information, see Item 4.B.“Business — Commercial Relationships.”
Costs of Revenue
Costs of revenue include primarily cost of raw materials, direct labor, contract manufacturing expenses, and in-bound and internal shipping and handling expenses.
Gross Profit (Loss) and Gross Margin
Gross margin reflects our gross profit divided by revenue.
Operating Expenses
Our operating expenses since inception have consisted of research and development expenses, sales and marketing expenses and general and administrative expenses.
Research and Development Expenses
The largest component of our total operating expenses has historically been our investment in research and development activities. We conduct our research and development primarily in-house, and we also contract with third-party vendors to conduct supplemental research, such as that related to the data generated by the INVU platform and certain of our strategic partners, and to assist with preparation of publications thereon. Research and development expenses consist mainly of costs incurred in connection with the research and development of our products and related clinical and regulatory activities. These expenses include:
| ● | employee-related expenses, including salaries, related benefits and share-based compensation expenses for employees engaged in research and development activities; |
139
| ● | expenses incurred in connection with the development of our products, including payments made pursuant to agreements with third parties, such as outside consultants related to development process and manufacturing activities; |
| ● | costs of components and materials; |
| ● | costs of external testing facility; |
| ● | facilities, depreciation and other expenses, including direct or allocated expenses for rent and maintenance of facilities, as well as insurance costs; |
| ● | costs related to compliance with regulatory requirements; and |
| ● | expenses related to clinical activities. |
We recognize research and development expenses to the statement of operations as they are incurred. Research and development activities are central to our business. We expect that our research and development expenses will be consistent over the next several years as we continue developing various aspects of our INVU platform, including without limitation: hardware, algorithmic engines, machine learning and AI modules, cloud-based infrastructure and product user experience/user interface, or UX/UI design.
At this time, we cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary to complete all the future development of our products. This uncertainty is due to the numerous risks and uncertainties associated with product development, including the uncertainty of:
| ● | the timing and progress of development activities; |
| ● | our ability to maintain our current research and development programs and to establish new ones; |
| ● | the receipt of regulatory approvals from applicable regulatory authorities; |
| ● | the timing, receipt and terms of any marketing clearances and approvals from applicable regulatory authorities; |
| ● | our ability to establish new licensing or collaboration arrangements; |
| ● | the performance of our future collaborators, if any; |
| ● | establishing and maintaining commercial manufacturing capabilities or making arrangements with third-party manufacturers; |
| ● | obtaining, maintaining, defending and enforcing patent claims and other intellectual property rights; |
| ● | the costs associated with compliance in the heavily regulated healthcare industry, changes to which could result in increased costs and/or reduced revenue, significantly and adversely affecting our business and future product development; and |
| ● | maintaining a continued acceptable safety profile of the products following approval. |
Any changes in the outcome of any of these variables with respect to the development of our products could result in a significant change in the costs and timing associated with the development of these products.
140
Sales and Marketing Expenses
Sales and marketing expenses consist primarily of salaries and related benefits and share-based compensation for employees engaged in sales and marketing activities, as well as public relations and marketing expenses, allocated expenses for rent and maintenance of facilities and insurance costs.
We expect that our sales and marketing expenses will increase as we expand our sales, marketing and sales support teams and increase sales and marketing activity in the United States, as we attempt to accelerate adoption and commercialization of our INVU platform. We expect that the staff growth will also increase share-based compensation expenses.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related benefits and share-based compensation, as well as other expenses, direct or allocated, for rent, maintenance of facilities, utilities, insurance and professional fees for legal, IP, consulting, accounting and audit services.
We expect that our general and administrative expenses will increase as a result of our planned growth and while operating as a public company, including expenses related to SEC compliance and Nasdaq listing, additional insurance, investor relations activities and the need for additional administrative and professional services, such as accounting, legal, regulatory and tax.
We also expect our administrative expenses, including share-based compensation expenses, to increase as we increase our headcount, expand our facilities and enhance our information technology to support our operations as a public company.
During the years ended December 31, 2023, 2022, and 2021, general and administrative expenses also included certain accrued expenses derived from our obligation to pay any taxes resulting from the exercise of certain options granted to our former Chief Innovation Officer and any taxes resulting from the sale from the shares underlying such options.
Financial Income (Expenses), Net
Financial income (expenses) consists primarily of the exchange rate difference between U.S. dollars and NIS, as well as commissions paid in connection with our financing activities during the years ended December 31, 2023, 2022, and 2021.
For the years ended December 31, 2023 and 2022, financial expenses consisted primarily of fundraising costs and commissions paid in connection with SAFE investments.
Provision for Income Taxes
Since our inception, we have not recorded any tax benefits for the net losses we incurred in any year due to the uncertainty of realizing a benefit from those losses. As of December 31, 2023, we had a net operating loss carryforward of $(76,689) for which a full valuation allowance was provided.
We account for uncertain tax positions in accordance with ASC 740-10. ASC 740-10 contains a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than not that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of any related appeals or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50% (cumulative probability) likely to be realized upon ultimate settlement.
We have recognized a full valuation allowance in respect of deferred income tax assets.
141
Recently Issued and Adopted Accounting Standards
See Note 2 to Nuvo’s audited consolidated financial statements included elsewhere in this Annual Report for more information.
Emerging Growth Company Status
The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company.” The JOBS Act permits an “emerging growth company” to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. Such companies may use this extended transition period under the JOBS Act until the earlier of the date it (i) is no longer an emerging growth company or (ii) affirmatively and irrevocably opts out of the extended transition period provided in the JOBS Act. As a result, the financial statements of an “emerging growth company” may not be comparable to companies who have adopted new or revised accounting pronouncements.
Following the Business Combination, Holdco will remain an emerging growth company until the earlier of (i) the last day of the fiscal year (a) following the fifth anniversary of the completion of the Closing, (b) in which Holdco has total annual gross revenue of at least $1.235 billion or (c) in which Holdco is deemed to be a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, and (ii) the date on which Holdco has issued more than $1.00 billion in non-convertible debt during the prior three-year period.
142
Results of Operations
Results of Operations for the years ended December 31, 2023 and 2022
The following table summarizes our results of operations for the years ended December 31, 2023 and 2022.
|
Year Ended December 31, |
Change | |||||||||||||||
| 2023 | 2022 | $ | % | |||||||||||||
| (dollars in thousands) | ||||||||||||||||
| Revenues | $ | 176 | $ | - | $ | 176 | NM | |||||||||
| Cost of revenues | 191 | - | 191 | NM | ||||||||||||
| GROSS LOSS | (15 | ) | - | (15 | ) | NM | ||||||||||
| Operating expenses | ||||||||||||||||
| Research and development, net | 8,324 | 9,893 | (1,569 | ) | (15.9 | )% | ||||||||||
| Sales and marketing | 3,221 | 4,752 | (1,531 | ) | (32.2 | )% | ||||||||||
| General and administrative | 5,073 | 6,161 | (1,088 | ) | (17.7 | )% | ||||||||||
| Total operating expenses | 16,618 | 20,806 | (4,188 | ) | (20.1 | )% | ||||||||||
| LOSS FROM OPERATIONS | (16,633 | ) | (20,806 | ) | 4,173 | (20.1 | )% | |||||||||
| Change in fair value of financial instruments | (18,017 | ) | 971 | (18,988 | ) | (1,955.5 | )% | |||||||||
| Other financial expenses, net | (44 | ) | (69 | ) | 25 | (36.2 | )% | |||||||||
| LOSS BEFORE TAX EXPENSE (BENEFIT) | (34,694 | ) | (19,904 | ) | (14,790 | ) | 74.3 | % | ||||||||
| TAX EXPENSES (BENEFIT) | (1,039 | ) | 775 | (1,814 | ) | (234.1 | )% | |||||||||
| TOTAL COMPREHENSIVE LOSS | $ | (33,655 | ) | $ | (20,679 | ) | $ | (12,976 | ) | 62.7 | % | |||||
NM denotes percentages that are not meaningful.
Research and Development Expenses
The table below summarizes our research and development expenses incurred during the years presented:
|
Year Ended December 31, |
Change | |||||||||||||||
| 2023 | 2022 | $ | % | |||||||||||||
| (dollars in thousands) | ||||||||||||||||
| Research and Development Expenses: | ||||||||||||||||
| Salaries and wages | $ | 4,734 | $ | 5,557 | $ | (823 | ) | (14.8 | )% | |||||||
| Share-based compensation | 1,346 | 1,664 | (318 | ) | (19.1 | )% | ||||||||||
| Rent, office and utilities, software licenses and communication | 1,740 | 1,834 | (94 | ) | (5.1 | )% | ||||||||||
| Professional services | 486 | 556 | (70 | ) | (12.6 | )% | ||||||||||
| Other | 18 | 359 | (341 | ) | (95.0 | )% | ||||||||||
| Research and development, gross | $ | 8,324 | $ | 9,970 | $ | (1,646 | ) | (16.5 | )% | |||||||
| Less - participation of R&D expenses | - | (77 | ) | 77 | (100.0 | )% | ||||||||||
| Total research and development, net | $ | 8,324 | $ | 9,893 | $ | (1,569 | ) | (15.9 | )% | |||||||
Research and development expenses decreased by $1.6 million, or 15.9%, during the year ended December 31, 2023 compared to the year ended December 31, 2022. The decrease was mainly attributable to a decrease in salaries and wages of $0.8 million, a decrease in share-based compensation of $0.3 million, and a decrease in other expenses of $0.3 million. The overall decrease in expenses was part of our cost cutting initiative deployed to adjust our operations to the challenging fundraising environment during 2023 and 2022.
143
Sales and Marketing Expenses
The table below summarizes our sales and marketing expenses incurred during the years presented:
|
Year Ended December 31, |
Change | |||||||||||||||
| 2023 | 2022 | $ | % | |||||||||||||
| (dollars in thousands) | ||||||||||||||||
| Sales and Marketing Expenses: | ||||||||||||||||
| Salaries and wages | $ | 2,130 | $ | 2,456 | $ | (326 | ) | (13.3 | )% | |||||||
| Share-based compensation | 508 | 1,787 | (1,279 | ) | (71.6 | )% | ||||||||||
| Marketing and business development | 583 | 509 | 74 | 14.5 | % | |||||||||||
| Total sales and marketing expenses | $ | 3,221 | $ | 4,752 | $ | (1,531 | ) | (32.2 | )% | |||||||
Sales and marketing expenses decreased by $1.5 million, or 32.2%, during the year ended December 31, 2023 compared to the year ended December 31, 2022. The decrease was primarily attributable to a decrease of $1.3 million in share-based compensation expense and a decrease of $0.3 million in salaries and wages, due to cost cutting initiatives deployed to adjust our operations to the challenging fundraising environment during 2023 and 2022.
General and Administrative Expenses
The table below summarizes our general and administrative expenses incurred during the years presented:
|
Year Ended December 31, |
Change | |||||||||||||||
| 2023 | 2022 | $ | % | |||||||||||||
| General and Administrative Expenses: | ||||||||||||||||
| Salaries and wages | $ | 1,039 | $ | 1,508 | $ | (469 | ) | (31.1 | )% | |||||||
| Share-based compensation | 1,241 | 4,323 | (3,082 | ) | (71.3 | )% | ||||||||||
| Change in fair value of commitment to shareholder | (1,036 | ) | (1,500 | ) | 464 | (30.9 | )% | |||||||||
| Rent, office and utilities, software license and communication | 48 | 1,079 | (1,031 | ) | (95.6 | )% | ||||||||||
| Professional services | 3,747 | 441 | 3,306 | 749.7 | % | |||||||||||
| Other | 34 | 310 | (276 | ) | (89.0 | )% | ||||||||||
| Total general and administrative expenses | $ | 5,073 | $ | 6,161 | $ | (1,088 | ) | (17.7 | )% | |||||||
General and administrative expenses decreased by $1.1 million, or 17.7%, during the year ended December 31, 2023 compared to the year ended December 31, 2022. The decrease was primarily attributable to a decrease of $3.1 million in share-based compensation expense, a decrease of $0.5 million in salaries and wages, and $1.0 million in rent, office, utilities, software licenses, and communication, and a decrease of $0.3 million in other expenses due to cost cutting initiatives deployed to adjust our operations to the challenging fund-raising environment during 2023 and 2022. The decreases were partially offset by an increase of $3.3 million professional services during the year ended December 31, 2023 incurred in connection with the pending Business Combination as well as an increase of $0.5 million in change in fair value of commitment to shareholder calculated based on the decrease in the Company’s 409A valuation.
Operating Loss
For the year ended December 31, 2023, our operating loss decreased by $4.2 million, or 20.1%, from $20.1 million during the year ended December 31, 2022 to $16.6 million during the year ended December 31, 2023. This reduction is mainly driven by a reduction in work-force as well as other general cost cutting initiatives taken in light of a difficult funding environment during 2023 and 2022.
144
Change in Fair Value of Financial Instruments
The gain of $1.0 million from the change in fair value of financial instruments during the year ended December 31, 2022 decreased by $19.0 million to a loss of $18.0 million during the year ended December 31, 2023 due to the decline in the Company’s valuation applied to the Nuvo Crossover Preferred Shares and SAFE financial instruments recorded on the Company’s balance sheet.
Other Financial Expenses, Net
Other financial expenses, net decreased by $25 thousand, or 36.2% from $69 thousand during the year ended December 31, 2022 to $44 thousand during the year ended December 31, 2023.
Tax expenses (benefit)
Tax expenses (benefit) increased by $1.8 million, or 234.1%, from a tax expense of $0.8 million during the year ended December 31, 2022 to a tax benefit of $1.0 million during the year ended December 31, 2023, due to a reversal of a portion of the Company’s uncertain tax position liabilities.
145
Results of Operations
Results of Operations for the years ended December 31, 2022 and 2021
The following table summarizes our results of operations for the years ended December 31, 2022 and 2021.
|
Year Ended December 31, |
Change | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| (dollars in thousands) | ||||||||||||||||
| Operating expenses | ||||||||||||||||
| Research and development, net | $ | 9,893 | $ | 10,470 | $ | (577 | ) | (5.5 | )% | |||||||
| Sales and marketing | 4,752 | 2,369 | 2,383 | 100.6 | % | |||||||||||
| General and administrative | 6,161 | 14,727 | (8,566 | ) | (58.2 | )% | ||||||||||
| Total operating expenses | 20,806 | 27,566 | (6,760 | ) | (24.5 | )% | ||||||||||
| LOSS FROM OPERATIONS | (20,806 | ) | (27,566 | ) | 6,760 | (24.5 | )% | |||||||||
| Change in fair value of financial instruments | 971 | (5,948 | ) | 6,919 | (116.3 | )% | ||||||||||
| Other financial expenses, net | (69 | ) | (565 | ) | 496 | (87.8 | )% | |||||||||
| LOSS BEFORE TAX EXPENSE (BENEFIT) | (19,904 | ) | (34,079 | ) | 14,175 | (41.6 | )% | |||||||||
| TAX EXPENSES (BENEFIT) | 775 | 433 | 342 | 79.0 | % | |||||||||||
| TOTAL COMPREHENSIVE LOSS | $ | (20,679 | ) | $ | (34,512 | ) | $ | 13,833 | (40.1 | )% | ||||||
Research and Development Expenses
The table below summarizes our research and development expenses incurred during the years presented:
|
Year Ended December 31, |
Change | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| (dollars in thousands) | ||||||||||||||||
| Research and Development Expenses: | ||||||||||||||||
| Salaries and wages | $ | 5,557 | $ | 5,293 | $ | 264 | 5.0 | % | ||||||||
| Share-based compensation | 1,664 | 2,784 | (1,120 | ) | (40.2 | )% | ||||||||||
| Rent, office and utilities, software licenses and communication | 1,834 | 1,924 | (90 | ) | (4.7 | )% | ||||||||||
| Professional services | 556 | 215 | 341 | 158.6 | % | |||||||||||
| Other | 359 | 254 | 105 | 41.3 | % | |||||||||||
| Research and development, gross | $ | 9,970 | $ | 10,470 | $ | (500 | ) | (4.8 | )% | |||||||
| Less - participation of R&D expenses | (77 | ) | - | (77 | ) | NM | ||||||||||
| Total research and development, net | $ | 9,893 | $ | 10,470 | $ | (577 | ) | (5.5 | )% | |||||||
Research and development expenses decreased by $0.6 million, or 5.5%, in 2022 compared to 2021. The decrease was mainly attributable to a decrease in share-based compensation in the amount of $1.1 million during the year ended December 31, 2022 and a decrease in rent, office and utilities, software licenses and communication expenses of $0.1 million during that year. Such decrease in research and development expense was partially offset by increased salaries and wages of $0.3 million in 2022 compared to the prior year mainly attributable to growth in our Clinical and Regulatory team as well as an increase of $0.3 million in professional services expenses. The overall decrease in expenses was part of our cost cutting initiative deployed to adjust our operations to the challenging fundraising environment during 2022.
146
Sales and Marketing Expenses
The table below summarizes our sales and marketing expenses incurred during the years presented:
| Year Ended December 31, |
Change | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| (dollars in thousands) | ||||||||||||||||
| Sales and Marketing Expenses: | ||||||||||||||||
| Salaries and wages | $ | 2,456 | $ | 1,638 | $ | 818 | 49.9 | % | ||||||||
| Share-based compensation | 1,787 | 449 | 1,338 | 298.0 | % | |||||||||||
| Marketing and business development | 509 | 282 | 227 | 80.5 | % | |||||||||||
| Total sales and marketing expenses | $ | 4,752 | $ | 2,369 | $ | 2,383 | 100.6 | % | ||||||||
Sales and marketing expenses increased by $2.4 million, or 100.6%, in 2022 compared to 2021. This increase was primarily attributable to an increase in share-based compensation in the amount of $1.3 million and increase in salaries and wages of $0.8 million, which resulted from an increase in headcount of employees to support the growth of our marketing activities.
General and Administrative Expenses
The table below summarizes our general and administrative expenses incurred during the years presented:
|
Year Ended December 31, |
Change | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| General and Administrative Expenses: | ||||||||||||||||
| Salaries and wages | $ | 1,508 | $ | 1,300 | $ | 208 | 16.0 | % | ||||||||
| Share-based compensation | 4,323 | 6,517 | (2,194 | ) | (33.7 | )% | ||||||||||
| Change in fair value of commitment to shareholder | (1,500 | ) | 3,445 | (4,945 | ) | (143.5 | )% | |||||||||
| Rent, office and utilities, software license and communication | 1,079 | 597 | 482 | 80.7 | % | |||||||||||
| Professional services | 441 | 496 | (55 | ) | (11.1 | )% | ||||||||||
| Other | 310 | 2,372 | (2,062 | ) | (86.9 | )% | ||||||||||
| Total general and administrative expenses | $ | 6,161 | $ | 14,727 | $ | (8,566 | ) | (58.2 | )% | |||||||
General and administrative expenses decreased by $8.6 million, or 58.2%, in 2022 compared to 2021. The decrease was primarily attributable to a decrease of $4.9 million in a commitment to a shareholder of Nuvo resulting from an agreement with our founder and former Chief Executive Officer as well as a decrease of $2.2 million in share-based compensation. This agreement was entered into as part of the termination of the founder’s employment agreement with the Company. Additionally, there was a decrease of $2.1 million in other expenses related to one-time fees related to our public filings in 2021 in connection with a potential initial public offering. The reduction in general and administrative expenses was partially offset by an increase of $0.2 million in salaries and wages, which was mainly due to the salary to our then-new Chief Executive Officer as well as an increase of $0.5 million in rent, office and utilities, software license and communication.
Operating Loss
For the year ended December 31, 2022 our operating loss decreased to $20.8 million from $27.6 million in the prior year. This decrease of $6.8 million represents a 24.5% reduction in operating expenses for the year ended December 31, 2022 over the prior year. This reduction is mainly driven by one-off expenses associated with the Company’s public filings in 2021 in connection with a potential initial public offering, as well as cost cutting initiatives taken in light of a difficult funding environment during the 2022 calendar year.
Financial Expenses, Net
Financial expenses, net in 2022 were mainly derived from $0.2 million of bank commission related to exchanging funds from U.S. dollars to NIS. These expenses were partly offset by favorable movement in the exchange rate between the NIS and the U.S. dollar because most of our cash is denominated in U.S. dollars while most of our employees are paid in NIS.
147
| B. | Liquidity and Capital Resources |
Sources of Liquidity
Since our inception and through December 31, 2022, we had not generated any revenue from product sales or otherwise and have incurred significant operating losses and negative cash flows from operations. During the year ended December 31, 2023, we began to generate revenue. However, we continue to incur signification operating losses and negative cash flows from operations. During the years ended December 31, 2023, 2022, and 2021, we incurred net losses of $33.7 million, $20.7 million and $34.5 million, respectively. As of December 31, 2023, we had an accumulated deficit of approximately $143.8 million and working capital, which is defined as current assets minus current liabilities, of approximately $(29.4) million. As of December 31, 2023, our primary sources of liquidity were cash and cash equivalents totaling $0.6 million. We expect to incur additional losses and operating expenses in future periods. As we hire incremental sales and marketing personnel and focus resources on building the commercial aspects of our business, we expect to continue to incur significant research and development expenses associated with moving our current product offering forward, including personnel related expenses and costs of conducting preclinical studies and clinical trials. We expect that general and administrative expenses will also increase as we expand our finance and administrative staff in connection with our transition to a public company. We have funded our operations to date primarily with proceeds from the sale of our ordinary shares, sale of our redeemable crossover preferred shares, SAFEs, Convertible Loans and Bridge Loans. Our future funding needs and related risks are discussed in further detail under “— Funding Requirements” below.
From June 2020 through the year ended December 31, 2023, we entered into certain Simple Agreements for Future Equity (the “Nuvo SAFEs”), which are characterized as liabilities, with several existing shareholders and new investors.
The Nuvo SAFEs were divided into three types:
| 1. | SAFEs entered into prior to April 26, 2021, for approximately $15 million, which provided for conversion of the respective SAFE at a price per share representing the lower of (a) $200 million pre-money valuation cap; or (b) a 15% discount rate on the price per share paid by the investors at the future financing round (whichever calculation results in the issuance of the greater number of shares to the SAFE holder); |
| 2. | SAFEs entered into on or after April 26, 2021 and before July 4, 2022 for approximately $8 million, which originally provided conversion of the respective SAFE at a price per share representing the lower of (a) a $625 million pre-money valuation cap; or (b) a 25% discount rate on the price per share paid by the investors at the future financing round (whichever calculation results in the issuance of the greater number of shares to the SAFE holder). All of these SAFEs, except SAFEs representing investment of $0.2 million, have been amended to provide for a $400 million pre-money valuation cap (instead of $625 million); and |
| 3. | SAFEs entered into on or after May 29, 2022 in connection with the entry into the Nuvo Convertible Loans (as defined and described below), which provide conversion of the respective SAFE at a price per share representing the lower of (a) a $350 million pre-money valuation cap; or (b) a 25% discount rate on the price per share paid by the investors at the future financing round (whichever calculation results in the issuance of the greater number of shares to the SAFE holder). |
The Nuvo SAFEs contain certain triggering events which provide for the conversion of the Nuvo SAFEs into shares as follows: (i) an equity financing in which the Company issues and sells shares for an aggregate consideration of at least $20 million or, with respect to the Nuvo SAFEs described in section 3 above, $15 million (“Equity Financing”) or (ii) either a change of control transaction or an initial public offering, whichever occurs sooner, which in each case is referred to as a “Liquidity Event.” Upon the occurrence of a Liquidity Event, the respective SAFE investor will, at its discretion, receive either a cash payment or shares of the then existing most senior series, which conversion into shares will be based on a conversion price per share based on the pre-money valuation cap of the respective Nuvo SAFE, or, in the case of the Nuvo SAFEs listed in section 3 above, the amount received by either the Company or its shareholders multiplied by 75%, divided by our outstanding capitalization in effect immediately prior to the Liquidity Event, calculated on an as-converted and fully diluted basis. In addition, given that a Liquidity Event, such as a change of control transaction, is not at our determination, such Nuvo SAFEs are characterized as liabilities. In connection with the entry into the Business Combination Agreement, the SAFEs were amended as described in further detail below.
148
From May 29, 2022 through the year ended December 31, 2022, Nuvo entered into several loan agreements (the “Nuvo Convertible Loans”) with certain investors, representing an aggregate principal amount of approximately $7.4 million, out of which an aggregate principal amount of $2.4 million was lent to Nuvo by related parties. The Nuvo Convertible Loans bear interest at a rate of 2% per month, payable at the maturity date (unless the holder elects to have any portion of the interest applied to the SAFEs as described below), and mature 12 months from the date of the applicable Nuvo Convertible Loan agreement, which maturity can be extended at Nuvo’s option by an additional 12 months. If Nuvo elects to extend the maturity date of a Nuvo Convertible Loan, the applicable lender shall receive a one-time extension fee equal to 20% of the loan principal amount, which shall be applied to the “purchase amount” of the SAFE issued to each Nuvo Convertible Loan lender (the “Extension Fee”). The Nuvo Convertible Loans may be prepaid by Nuvo in whole or in part at any time without prepayment penalty.
As an incentive to provide the Nuvo Convertible Loans, each Nuvo Convertible Loan investor received a SAFE in connection with entry into the Nuvo Convertible Loan agreement, representing a SAFE “purchase amount” equal to 20% of such respective investor’s Nuvo Convertible Loan’s principal loan amount, which purchase amount may be increased by (i) any amount of the Nuvo Convertible Loan’s principal and/or any accrued and unpaid interest thereon at the investor’s option and (ii) the Extension Fee.
Upon the occurrence of an equity investment in Nuvo in the aggregate amount of at least $15 million, Nuvo shall repay the outstanding principal and accrued but unpaid interest on the Nuvo Convertible Loans, unless an investor has exercised its option to convert the Nuvo Convertible Loan’s principal and/or interest into the related SAFE’s purchase amount.
From August through October 2023, the Company signed several agreements to issue Nuvo Crossover Preferred Shares at a per share issuance price of $7.0265 for total proceeds of $13.0 million. Upon the consummation of a de-SPAC transaction, the Nuvo Crossover Preferred Shares were converted to Holdco preferred shares with the same rights associated to those shares of Holdco.
In August and September 2023, Nuvo obtained the necessary corporate consents for the amendment of the Nuvo SAFEs (the “Nuvo SAFE Amendment”), which was intended, inter alia, (a) to equalize the economic conversion terms across the different types of the Nuvo SAFEs described above, such that: (1) the discount rate in all Nuvo SAFEs shall be 25% and (2) the pre-money valuation cap in all Nuvo SAFEs shall be $200 million; and (b) to set the conversion terms of the Nuvo SAFEs in connection with the consummation of the Business Combination (which shall not otherwise constitute a Liquidity Event pursuant to the terms of the Nuvo SAFE Amendment), such that, upon the consummation of the Business Combination, the Nuvo SAFEs will automatically convert into Nuvo Shares based on a price per share representing the lower of (1) a $150 million pre-money valuation cap, or (2) a 25% discount on the price per share imputed to the Nuvo Shares pursuant to the Business Combination Agreement (whichever results in the issuance to the Nuvo SAFE holder of a greater number of Nuvo Shares). Accordingly, at the Closing, Nuvo issued approximately 3.56 million Nuvo Shares in satisfaction and discharge of its obligations under the Nuvo SAFEs, in accordance with the provisions of the Nuvo SAFE Amendment.
In August and September 2023, Nuvo obtained the necessary corporate consents for the Nuvo Loan Amendment, such that, in exchange for the Extension Fee under the original loan terms, the maturity date of each Nuvo Convertible Loan was extended to the earlier of the second anniversary of the applicable loan or the Closing. In addition, pursuant to the Nuvo Loan Amendment, each lender has agreed to apply the principal amount of the Nuvo Convertible Loan, the accrued and unpaid interest thereon and the Extension Fee to the purchase amount of the related Nuvo SAFE described in section 3 above. As such, in connection with the Closing, Nuvo’s repayment obligations under the Nuvo Convertible Loans converted to an aggregate SAFE purchase amount of approximately $12.25 million, which then converted, pursuant to the terms of the Nuvo SAFE Amendment, into approximately 1.89 million Nuvo Shares which was exchanged for Holdco Shares pursuant to the terms of the Business Combination Agreement.
149
In November and December 2023, the Company entered into several Bridge Loan agreements with respect to Bridge Financing Notes and received cash from certain investors, representing an aggregate principal amount received of $2.050 million, out of which $0.4 million was lent to Nuvo by related parties. The Nuvo Bridge Notes bear interest at a rate of 15% per annum with a maturity date of 12 months from issuance date, the closing of the de-SPAC or IPO, or the closing of a Qualified Financing. While interest will be payable in cash at the maturity date, investors may choose to receive the principal amount in cash or to convert the Principal Amount into ordinary shares of the Company at a price per share of $7.0265. Bridge Financing Notes executed by certain investors for the cash received by December 31, 2023 provide for a total of 1,112,930 warrants, of which 796,938 and 315,947 will expire on the 3-year anniversary and 4-year anniversary of the issuance date, respectively. Of the 1,112,930 total warrants, 113,855 were due to related parties.
As of the date hereof, approximately $8.5245 million in principal amount of Bridge Financing Notes has been received by Nuvo.
From March 24, 2024 through April 8, 2024, Nuvo entered into amendments to all of the existing Bridge Financing Notes representing $6.5732 million principal amount of the Bridge Financing Notes, to extend the maturity dates thereof (the “Bridge Financing Notes Amendments”). All new Bridge Financing Notes since April 8, 2024 include the amended maturity definition. Prior to the Bridge Financing Notes Amendments, the Bridge Financing Notes were scheduled to mature on the earlier of (i) twelve months from the issuance date thereof, (ii) the closing of the Business Combination, (iii) the closing of an initial public offering, or (iv) the closing of a bona fide financing by Nuvo for the principal purpose of raising capital, through the sale of Nuvo securities in whatever form or type (whether debt or equity) that raises in excess of $10,000,000 in gross proceeds. Pursuant to the Bridge Financing Notes Amendments, the maturity date of the amended Bridge Financing Notes was revised to be the earlier of (i) twelve months from the issuance date thereof, (ii) six (6) months following the closing of the Business Combination, (iii) six (6) months following the closing of an initial public offering, or (iv) the closing of a bona fide financing by Nuvo for the principal purpose of raising capital, through the sale of Nuvo securities in whatever form or type (whether debt or equity) that raises in excess of $25,000,000 in gross proceeds.
Each Bridge Financing Note is secured by all of Nuvo’s intellectual property, and Nuvo has filed collateral assignments/financing statements with the United States Patent & Trademark Office and is in the process of filing collateral assignments/financing statements with Nuvo’s Registrar in Israel. Gaingels 10x Capital Diversity Fund I, LP, a Bridge Financing Holder and an affiliate of a member of the Sponsor serves as collateral agent with respect to the collateral securing the Bridge Financing Notes. Upon the occurrence of any event of default described therein, the outstanding balance under the Bridge Financing Notes shall become immediately due and payable upon election of the Bridge Financing Holder and following a written demand notice sent to Nuvo.
In consideration for the services to be rendered under certain advisory services agreements between the Bridge Financing Holders and Nuvo, Nuvo issued a warrant to each Bridge Financing Holder, whereby the Bridge Financing Holder is given the right to purchase such number of Nuvo Shares (or, post-Closing, Holdco Ordinary Shares after applying the equity exchange ratio of 96.139%) equal to (2x) the principal amount of the Holder’s Bridge Financing Note divided by the same price per share noted above (i.e., $7.0265), at an exercise price of NIS 0.01.
The initial agreement with a certain investor to obtain bridge loan financing in the amount of $1.0 million required, as a condition of the funding, the Company to obtain $2.0 million of additional financing from third parties within 30 days of the issuance of the note. The Company met this condition in January 2024.
Cash Flows
The following table summarizes our cash flows for the periods indicated:
|
Year Ended December 31, |
||||||||
| 2023 | 2022 | |||||||
| (dollars in thousands) | ||||||||
| Net cash provided by (used in): | ||||||||
| Operating activities | $ | (14,956 | ) | $ | (13,471 | ) | ||
| Investing activities | (38 | ) | (277 | ) | ||||
| Financing activities | 14,444 | 9,825 | ||||||
| Net decrease in cash, cash equivalents, and restricted cash | $ | (550 | ) | $ | (3,923 | ) | ||
150
Net Cash Used in Operating Activities
Net cash used in operating activities during the year ended December 31, 2023 was $15.0, and consisted primarily of our net loss of $33.7 million, partially offset by non-cash charges of $20.3 million. These non-cash charges consisted of $18.0 million resulting from remeasurement of financial instruments associated with the Nuvo Crossover Preferred Shares and SAFEs, $3.1 million of share-based compensation expense, and $0.2 million of depreciation and amortization, partially offset by $1.0 million change in fair value of commitment to shareholder. Please see Notes 2 and 9 of our audited financial statements included elsewhere in this Annual Report for more information on the accounting treatment of the SAFEs. The net cash outflows from changes in operating assets and liabilities were primarily the result of an increase of $0.6 million for other assets, a decrease of $0.3 million for convertible loans, and a decrease of $0.2 million for accrued severance pay, and a decrease of $0.4 million for other accounts payable.
Net cash used in operating activities during the year ended December 31, 2022 was $13.5 million, consisting primarily of our net loss of $20.7 million, partially offset by non-cash charges of $5.8 million. These non-cash charges consisted primarily of $7.8 million of share-based compensation, depreciation and amortization of $0.5 million, partially offset by a charge for the remeasurement of financial instruments of $1.0 million resulting from the SAFEs classification as a liability at fair value and adjustments made to their fair value at each reporting period, and by a $1.5 million change in fair value of commitment to shareholder. The net cash inflows from changes in operating assets and liabilities were primarily the result of an increase of $0.6 million for trade payables and $1.2 million of other accounts payable partially offset by an increase of $0.4 million of other current assets.
Net Cash Used in Investing Activities
Net cash used in investing activities for the year ended December 31, 2023 was $38 thousand, due to the purchase of computers and payments for production equipment.
Net cash used in investing activities for the year ended December 31, 2022 was $0.3 million, due to the purchase of computers and payments for production equipment.
Net Cash Used in Financing Activities
Net cash provided by financing activities for the year ended December 31, 2023 was $14.4 million, consisting primarily of $13.0 million proceeds from the issuance of Nuvo Crossover Preferred Shares, $2.0 million of proceeds from the issuance of bridge loan and warrants, and $0.5 million proceeds from the issuance of our Nuvo Convertible Loans. This was partially offset by $1.1 million repayment of convertible loans during the year ended December 31, 2023.
Net cash provided by financing activities during the year ended December 31, 2022 was $9.8 million, consisting primarily of $7.4 million of proceeds from the issuance of our Nuvo Convertible Loans and $2.4 million of proceeds from the issuance of SAFEs, net of issuance costs.
Funding Requirements
Because of the numerous risks and uncertainties associated with manufacture, research, development and commercialization of products, we are unable to estimate the exact amount of our capital requirements. Our future funding requirements will depend on, and could increase significantly as a result of, many factors, including:
| ● | the scope, progress, results and costs of researching and developing our INVU platform; |
| ● | the costs, timing and outcome of regulatory approval for additional features of our INVU platform and any future products, or for marketing authorization for any new countries or markets; |
151
| ● | the costs of future activities, including product sales, medical affairs, marketing, manufacturing and distribution, for our INVU platform; |
| ● | commercial manufacturing, shipping, and distribution of our products and sufficient inventory to support commercial launch; |
| ● | the scope, progress and costs of developing a sales and marketing network in the United States; |
| ● | the cost and timing of hiring new employees to support our continued growth; |
| ● | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; |
| ● | the ability to establish and maintain collaborations on favorable terms, if at all; |
| ● | the timing, receipt and amount of sales of our INVU platform, if any; |
| ● | our success in expanding and developing our operational, financial and management systems; |
| ● | our success at and the cost of becoming a public company; and |
| ● | the success and cost of our product acquisition activities, if any. |
A change in any of these or other variables with respect to our business or our INVU platform or any other product could significantly change the costs and timing associated with the development of such product. We expect our expenses to increase in connection with our ongoing activities and operations as we begin to grow our business. In addition, upon the closing of the Business Combination, we expect to incur additional costs as a result of operating as a public company. Further, our operating plans may change in the future, and we may need additional funds to meet operational needs and capital requirements associated with such operating plans.
Our primary uses of cash are to fund our operations as we continue to grow our business. We expect to continue to incur operating losses in the near term as our operating expenses will be increased to support the commercial growth of our business and as we incur the costs of becoming and operating as a public company. We expect that our sales and general and administrative expenses will continue to increase, and our research and development expenses will continue steady as we seek additional regulatory clearances, increase our INVU manufacturing volume, expand our marketing efforts, continue our research and development efforts and further develop INVU.
We expect that our near- and longer-term liquidity requirements will continue to consist of working capital and general expenses associated with the growth of our business. Depending on any new business models we might develop to monetize our solution, it is possible that we will be required to invest substantial resources in capital expenditures and fixed assets in both the short and long term.
Based on our current planned operations, we expect that our existing cash, proceeds from the closing of the Business Combination and anticipated net proceeds from the Bridge Loan program will enable us to fund our operating expenses for at least the next three months from the date of closing the Business Combination. We expect to execute a financing during the next three months to generate proceeds to provide cash to fund our operations, the amount of which cannot be estimated as of the date of the closing of the Business Combination. There can be no assurance that the financing will be consummated prior to the expiration of such three-month period, if at all, or that we will receive any such net proceeds in connection with the financing. Even if such financing is available, there is no assurance that it is obtainable on terms acceptable to us. In the absence of the net proceeds from the transactions, we will need additional financial support through the private raise of equity or other capital sources, or we will have to significantly reduce our expenditures, delay clinical trials, or enter into collaborations and/or licensing arrangements, in order to sustain operations for the next 12 months. We have concluded that this circumstance raises substantial doubt about our ability to continue as a going concern for at least one year from the date our financial statements were available for issuance. See Note 1 to Nuvo’s audited consolidated financial statements included elsewhere in this Annual Report, for additional information. Similarly, our independent registered public accounting firm included an explanatory paragraph in its report on Nuvo’s audited consolidated financial statements included in this Annual Report, describing the existence of substantial doubt about our ability to continue as a going concern.
152
We have based our estimates as to how long we expect we will be able to fund our operations on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect, in which case we would be required to obtain additional financing sooner than currently projected, which may not be available to us on acceptable terms, or at all. Our failure to raise capital as and when needed would have a negative impact on our financial condition and our ability to pursue our business strategy. We may raise additional capital through equity offerings, or other capital sources, including potentially, collaborations, licenses and other similar arrangements. If we do raise additional capital through public or private equity offerings, the ownership interest of our existing shareholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our existing shareholders’ rights. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise funds through collaborations, strategic partnerships or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or products or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds when needed, we may be required to delay, reduce or eliminate our product development or future commercialization efforts, or grant rights to develop and market our INVU platform that we would otherwise prefer to develop and market ourselves.
Contractual Obligations and Other Commitments
The following table summarizes our contractual obligations and other commitments as of December 31, 2023 and the effects that such obligations are expected to have on our liquidity and cash flows in future periods:
| Payments Due by Period | ||||||||||||
| Less than 1 Year |
1 to 3 Years | Total | ||||||||||
| Operating lease obligations | $ | 348 | $ | - | $ | 348 | ||||||
| Total | $ | 348 | $ | - | $ | 348 | ||||||
In December 2022, we entered into a new operating lease agreement in Tel Aviv. The monthly average rent expenses were approximately $32,000. The first lease period was for six months with an extension option for an additional six months with a monthly expense of approximately $30,000. In June 2023, we entered into a modified lease agreement exercising the above option ending on December 24, 2023 and adding an additional lease year ending on December 24, 2024, with a monthly expense of approximately $29,000, and a three-month termination option starting on March 30, 2024. To date, the Company has not exercised the termination option.
We have entered into contracts in the normal course of business with third parties. These contracts do not contain any minimum purchase commitments and are cancellable by us upon prior notice and, as a result, are not included in the table of contractual obligations and commitments above. Payments due upon cancellation consist only of payments for services provided and expenses incurred, including non-cancellable obligations of our service providers, up to the date of cancellation.
We are required to pay royalties to the State of Israel through the IIA, computed on the basis of proceeds from the sale or license of products, the development of which was supported by state grants. In accordance with the terms of the financial participation, the IIA is entitled to royalties on the sale or license of any product which development was supported with State of Israel participation. These royalties are generally 3% - 3.5% of sales until repayment of 100% of the grants (linked to the dollar) received by us plus annual interest at the SOFR rate.
The aggregate contingent obligation payable by us as of December 31, 2023 was approximately $1,164 million, which represents the gross amount of grants received by us from the IIA for two grant programs during the period from July 2014 to June 2016, including accrued interest as of December 31, 2023. As of December 31, 2023 we had not paid any royalties to the IIA.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
153
| C. | Research and Development, Patents and Licenses, Etc. |
For our research and development efforts, see Item 4.B. “Business Overview.” For information regarding our patents and proprietary rights, see Item 4.B. “Business Overview––Intellectual Property”.
| D. | Trend Information |
We are a women’s health and connected pregnancy care company, which recently began commercialization of our product, the INVU platform, and it is not possible for us to predict with any degree of accuracy the outcome of our commercialization efforts. As such, it is not possible for us to predict with any degree of accuracy any known trends, uncertainties, demands, commitments or events that are reasonably likely to have a material effect on our financial condition, including our liquidity and capital resources, or that would cause reported financial information to not necessarily be indicative of future operating results or financial conditions. Our results of operations and financial condition may be affected by various trends and factors discussed in Item 3.D. “Risk Factors,” Item 4 “Information on The Company” and elsewhere in this Item 5 “Operating and Financial Review and Prospects.”
| E. | Critical Accounting Estimates |
Our management’s discussion and analysis of our financial condition and results of operations is based on Nuvo’s financial statements included elsewhere in this Annual Report, that have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported income generated, and expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material.
While our significant accounting policies are more fully described in Note 2 to Nuvo’s audited financial statements included elsewhere in this Annual Report, we believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
We evaluate on an ongoing basis our assumptions, including those related to contingencies, income tax uncertainties, share-based compensation cost, fair value measurement of warrants, accretion of redeemable shares, and the fair value and useful life of intangible assets.
Inventories
Inventories are stated at the lower of cost or net realizable value. Inventory write-off is provided to cover risks arising from slow-moving items, technological obsolescence, excess inventories and discontinued products.
Inventory items are valued using the “average price” method. The Company assesses the carrying value of its inventory for each reporting period to ensure inventory is reported at the lower of cost or net realizable value. Charges for obsolete and slow-moving inventories are recorded based upon an analysis of specific identification of obsolete inventory items and quantification of slow-moving inventory items. These assessments consider various factors, technological obsolescence, estimated current and future market values and new product introduction. In cases when there is evidence that the anticipated utility of goods, in their disposal in the ordinary course of business, will be less than the historical cost of the inventory, the Company recognizes the difference as a current period charge to earnings and carries the inventory at the reduced cost basis until it is sold or disposed of. As of December 31, 2023 and 2022 the inventory is comprised of raw material and components only.
154
Share-Based Compensation
The Company accounts for share-based compensation in accordance with ASC No. 718, “Compensation-Stock Compensation” (“ASC No. 718”). ASC No. 718 requires companies to estimate the fair value of equity-based payment awards on the grant date using the Black-Scholes-Merton option pricing model, which is the most appropriate fair value method for its options awards. The option-pricing model requires a number of assumptions, of which the most significant are the expected share price volatility and the expected option term. Expected volatility was calculated based upon similar companies in the market, until sufficient historical data will be available. The expected term of options granted is calculated based upon the simplified method until sufficient historical exercise data will support using expected life assumptions. The risk-free interest rate is based on the yield from U.S. treasury bonds with an equivalent term to the expected life of the options. The Company has historically not paid dividends and has no foreseeable plans to pay dividends.
The fair value of Ordinary Shares underlying the options has historically been determined by management and approved by the Company’s Board of Directors. Because there has been no public market for the Company’s Ordinary Shares, the management has determined fair value of an Ordinary Share at the time of grant of the option by considering a number of objective and subjective factors including financing investment rounds, operating and financial performance, the lack of liquidity of share capital and general and industry-specific economic outlook, amongst other factors. The fair value of the underlying Ordinary Shares will be determined by the management until such time as the Company’s Ordinary Shares are listed on an established stock exchange.
The estimated fair value of the Company’s Ordinary Shares is determined by management using the Hybrid Method for the years 2023, 2022 and 2021, with the assistance of a third-party valuation expert.
The Company recognizes compensation cost for options and share awards that have a graded vesting schedule and contain only a service condition on accelerated attribution method for the entire award. Forfeitures are accounted for as they occur. For options granted to non-employees, the expected life of the option used is the contractual term of each such option. All other assumptions used to calculate the grant date fair value are generally consistent with the assumptions used for options granted to employees.
For awards with performance condition vesting features, compensation cost is recorded if it is probable that the performance condition will be achieved. If the Company originally estimated that it was not probable that the performance condition would be satisfied, compensation cost would not have been recognized. If the Company later determines that it is probable that the performance condition will be satisfied, it will recognize a cumulative catch-up adjustment to reflect the portion of the employee’s requisite service that has been provided to date and will continue to recognize compensation cost over the remaining requisite service period. The Company determined that the performance conditions as described above are not probable, and therefore no compensation cost was recognized.
Determination of Fair Value of Financial Instruments
The Company accounts for financial instruments under Financial Accounting Standards Board (“FASB”) Accounting Standards Codification 820 (“ASC 820”), Fair Value Measurements. This statement defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles, and expands disclosures about fair value measurements. To increase consistency and comparability in fair value measurements, ASC 820 establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three levels as follows:
Level 1 - quoted prices (unadjusted) in active markets for identical assets or liabilities.
Level 2 - observable inputs other than Level 1, quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets and liabilities in markets that are not active, and model-derived prices whose inputs are observable or whose significant value drivers are observable.
Level 3 - assets and liabilities whose significant value drivers are unobservable.
155
Observable inputs are based on market data obtained from independent sources, while unobservable inputs are based on the Company’s market assumptions.
Unobservable inputs require significant management judgment or estimation. In some cases, the inputs used to measure an asset or liability may fall into different levels of the fair value hierarchy. In those instances, the fair value measurement is required to be classified using the lowest level of input that is significant to the fair value measurement. Such determination requires significant management judgment.
During the years ended December 31, 2023 and 2022, Nuvo issued the Nuvo Convertible Loans, which were classified as liabilities and measured at fair value on the issuance date, with changes in fair value recognized in the statements of comprehensive loss and disclosed in our audited financial statements included elsewhere in this Annual Report.
During the year ended December 31, 2023, the Company signed several agreements for the purchase of Nuvo Crossover Preferred Shares with both new and existing investors. The proceeds were received before the Nuvo Crossover Preferred Shares were issued and the Company determined that, until issuance, the amounts received represented a contingent forward to issue redeemable crossover preferred shares. The contingent forward was accounted for as a liability measured at fair value at each balance sheet date. Upon issuance of the Nuvo Crossover Preferred Shares, the contingent forward was reclassified to mezzanine equity in the Company’s consolidated balance sheet.
During the years ended December 31, 2022, 2021 and 2020, Nuvo entered into certain SAFE agreements and classified the SAFE as a liability measured at cost on the issuance date, with changes in accordance with Accounting Standards Codification 480, “Financial Instruments”, Nuvo accounts for a SAFE as a liability at fair value and adjusts the instrument to fair value at each reporting period. This liability is subject to re-measurement at each balance sheet date until a triggering event, equity financing or a liquidity/dissolution occurs, and any change in fair value is recognized in Nuvo’s statements of comprehensive loss. The carrying amounts of Nuvo’s other financial assets and liabilities, such as accounts payable, approximate fair value due to the short-term nature of these instruments.
156
| ITEM 6. | DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES |
| A. | Directors and Senior Management |
The following table sets forth certain information, as of the date of this Annual Report, concerning the persons who serve as Holdco’s directors and executive officers. The corporate address for Holdco’s directors and executive officers is 94 Yigal Alon St., Tel Aviv, Israel 6789155:
| Name | Age | Position(s) | ||
| Robert Powell | 68 | Chief Executive Officer, Director | ||
| Douglas Blankenship | 62 | Chief Financial Officer | ||
| Laurence Klein | 61 | Director | ||
| Gerald Ostrov | 74 | Director (Chair) | ||
| Christina Spade | 54 | Director | ||
| Adriana Machado | 55 | Director |
Robert Powell, Holdco’s Chief Executive Officer, joined Holdco as an officer in February 2024, and has served on Holdco’s Board of Directors since the Closing. Mr. Powell served as the Chief Executive Officer and Chairman of Management at Fresenius Medical Care from 2013 to 2022. Since 1997, Mr. Powell served in multiple roles at Fresenius Medical Care, including serving as Vice Chairman and a member of the management board for North America, a member of the management board for the Renal Therapies Group North America, Senior Vice President and President of Products & Hospital Group, President of the Dialysis Products Division and President of Renal Product Technologies. Mr. Powell has more than 25 years of experience in the healthcare industry. From 1978 to 1996, he held various positions within Baxter International Inc. USA, Biogen Inc. USA and Ergo Sciences Inc. USA. Mr. Powell received his Bachelor of Science degree from Mississippi College.
Mr. Powell’s qualifications to serve on our Board of Directors include his extensive experience in management positions in the healthcare industry.
Douglas Blankenship, Holdco’s Chief Financial Officer, joined Holdco in February 2024. Prior to that, Mr. Blankenship served as the Chief Financial Officer of Humacyte, Inc. (Nasdaq: HUMA) from 2018 to 2021 and as Chief Financial Officer of Dova Pharmaceuticals, Inc. from 2017-2018. Before joining Dova Pharmaceuticals, Mr. Blankenship served in various roles at Genentech, Inc. from 2008 to 2015. Mr. Blankenship has also worked at Amgen, Inc., Abgenix, Inc., Virage Logic Corporation, and a number of other companies. Mr. Blankenship received his Bachelor of Science in Business Administration from California Polytechnic State University, San Luis Obispo and his Master of Business Administration from The Wharton School, University of Pennsylvania.
Laurence Klein was appointed to Holdco’s Board of Directors upon consummation of the Business Combination. Mr. Klein has served as the President of Nalay Inc, a private equity investment firm, since 2003. Mr. Klein is the President and is currently serving on the board of directors of CTK Holdings Limited and currently serves as the Managing Partner of Innopark Management Ltd Partnership and the Managing Partner of Nuvo Investors LLC (NILLC). Mr. Klein received his Bachelor of Engineering degree from McGill University in Montreal, Canada.
Mr. Klein’s qualifications to serve on our Board include his experience as the founder and Chief Executive Officer of several startup companies and his experience authoring patents.
Gerald M. Ostrov was appointed to Holdco’s Board of Directors upon consummation of the Business Combination. Mr. Ostrov has forty years of senior management experience in healthcare, including several positions with Johnson & Johnson from 1991 to 2006, and as CEO of Bausch & Lomb from 2008 to 2010, where he was responsible for managing sales with some of their most prestigious brands in their fields. Mr. Ostrov has served as a member of the board of directors of Entera Bio Ltd. (Nasdaq: ENTX) since 2019. He also currently serves on the board of directors of several privately held companies. Mr. Ostrov received his Bachelor of Science in Engineering from Cornell University and his MBA from Harvard Business School.
Mr. Ostrov’s qualifications to serve on our Board include his extensive experience in management positions in the healthcare industry.
157
Christina Spade, was appointed to Holdco’s Board of Directors upon consummation of the Business Combination. Ms. Spade has spent her career in media and entertainment, with a focus on consumer platforms. In 2021 and 2022, she held several leadership positions with AMC Networks, including Chief Financial Officer, Chief Operating Officer, and Chief Executive Officer. She previously served as Chief Financial Officer for ViacomCBS (currently Paramount Global) from 2019 to 2020 and for CBS Corporation from 2018 to 2019, prior to its merger with Viacom Inc. Prior to this, Ms. Spade was at Showtime Networks Inc. (“SNI”) for 21 years where she served in various roles, including Chief Financial Officer from 2012 to 2018, and was instrumental in the successful scaling of the Showtime multi-platform distribution strategy during the advent of streaming. Additionally, she served as Senior Vice President, Business Operations of SNI from 2000 to 2012. She also was an audit professional and CPA with PricewaterhouseCoopers from 1991 to 1997 in various roles. Ms. Spade is currently a member of the board of directors for The Paley Center for Media, since November 2022. She also founded in 2010, and serves as president, of ATR Children’s Foundation, which is a non-profit organization which helps children in need. Ms. Spade previously was an executive member of the board of directors for the T. Howard Foundation from 2015 to 2022. Ms. Spade was honored as a 2017 WICT Wonder Woman and served as an executive mentor in WICT’s mentorship program.
Ms. Spade’s qualifications to serve on our Board of Directors include her extensive experience in the finance and the entertainment industry.
Adriana Machado, was appointed to Holdco’s Board of Directors upon consummation of the Business Combination. Ms. Machado is a celebrated women business leader in Latin America and an outspoken advocate in the impact economy space. She served as the President and CEO of GE Brazil from December 2011 to August 2013 and as the Vice President of Government Affairs & Policy for Latin America from August 2013 to July 2015. She founded the Briyah Institute, a Benefit Corporation (B Corp) that bridges innovation, practice and purpose to inspire leaders to transform organizations co-creating an impact economy in April 2018. Ms. Machado serves on the advisory board of Securitas Bio since January 2021, on the US board of 501(c)(3) Instituto DARA since September 2023, and has been a supporter of the Brain Health Project, an initiative aimed at preventing Alzheimer’s Disease by promoting brain health and slowing cognitive decline. She also serves as a strategic partner to the WMB Partners since 2020 by connecting purpose-driven companies with transformational leaders, geared towards delivering positive impact to all stakeholders. Ms. Machado has a B.A. in Political Science from Universidade de Brasilia (UnB).
Ms. Machado’s qualifications to serve on our Board of Directors include her extensive experience in management positions.
Family Relationships
Noah Klein, Nuvo’s Vice President of Business Development, and Ari Klein, one of Nuvo’s employees, are the sons of Laurence Klein, one of Holdco’s directors. There are no other family relationships between any of our officers or directors.
| B. | Compensation |
The following table presents in the aggregate all compensation we paid to all of our directors and senior management as a group for the year ended December 31, 2023. The table does not include any amounts we paid to reimburse any of such persons for costs incurred in providing us with services during this period.
All amounts reported in the table below reflect our cost, in thousands of U.S. dollars. Amounts paid in NIS are translated into U.S. dollars at the rate of NIS 3.687 = U.S. $1.00, based on the average representative rate of exchange between the NIS and the U.S. dollar as reported by the Bank of Israel during such period of time.
| Salary, bonuses and Related Benefits |
Pension, Retirement and Other Similar Benefits |
Share Based Compensation |
||||||||||
| All directors and senior management as a group, consisting of three persons as of December 31, 2023. | $ | 561,679 | $ | 0 | $ | 363,544 | ||||||
As of December 31, 2023, there were 3,618,819 options to purchase 3,618,819 Nuvo Shares outstanding under Nuvo’s 2015 Share Incentive Plan (the “Nuvo 2015 Plan”), at a weighted average exercise price of $6.30 per share. Following the Closing, such options are exercisable to purchase Holdco Ordinary Shares at an exercise price adjusted as determined by the Equity Exchange Ratio.
158
Post-Business Combination Share Incentive Plan
Prior to the Business Combination, the Board adopted the 2024 Share Incentive Plan (the “2024 Plan”), which provides for the grant of equity-based incentive awards to its employees, directors, office holders, service providers and consultants in order to incentivize them to increase their efforts on behalf of Holdco and to promote the success of the Holdco’s business. In addition to the 2024 Plan, Holdco adopted the 2024 Employee Share Purchase Plan (“ESPP”). The 2024 Plan and the ESPP became effective upon the closing of the Business Combination.
The purpose of the 2024 Plan and the ESPP is to attract and retain highly qualified personnel and to provide key employees with additional incentive to increase their efforts on behalf and in the best interest of Holdco and its subsidiaries by giving them the opportunity to acquire a proprietary interest in Holdco as an incentive for them to remain in the service of Holdco.
2024 Share Incentive Plan
Shares Available for Grants. The maximum number of Holdco Ordinary Shares available for issuance under the 2024 Plan is equal to the sum of (i) 4,939,811 Holdco Ordinary Shares (representing 15% of the Holdco Ordinary Shares outstanding immediately following the Closing (the “Initial Share Pool”), (ii) any shares subject to awards under the 2024 Plan which have expired, or were cancelled, terminated, forfeited or settled in cash in lieu of issuance of shares or became unexercisable without having been exercised and (iii) an annual increase on the first day of each year beginning in 2025 and on January 1st of each calendar year thereafter during the term of the 2024 Plan, equal to the lesser of (A) 2.0% of the total number of outstanding Holdco Ordinary Shares of the Company on the last day of the immediately preceding calendar year, on a fully diluted basis; and (B) such amount as determined by our board of directors if so determined prior to January 1 of a calendar year, provided that no more than the amount of Holdco Ordinary Shares in the Initial Share Pool may be issued upon the exercise of Incentive Stock Options. If permitted by our board of directors, shares tendered to pay the exercise price or withholding tax obligations with respect to an award granted under the 2024 Plan may again be available for issuance under the 2024 Plan. Our board of directors may also reduce the number of Holdco Ordinary Shares reserved and available for issuance under the 2024 Plan in its discretion.
Administration. Our board of directors, or a duly authorized committee of our board of directors, or the administrator, administer the 2024 Plan. Under the 2024 Plan, the administrator has the authority, subject to applicable law, to interpret the terms of the 2024 Plan and any award agreements or awards granted thereunder, designate recipients of awards, determine and amend the terms of awards, including the exercise price of an option award, the fair market value of an ordinary share, the time and vesting schedule applicable to an award or the method of payment for an award, accelerate or amend the vesting schedule applicable to an award, prescribe the forms of agreement for use under the 2024 Plan and take all other actions and make all other determinations necessary for the administration of the 2024 Plan.
The administrator also has the authority to approve the conversion, substitution, cancellation or suspension under and in accordance with the 2024 Plan of any or all awards or Holdco Ordinary Shares, and the authority to modify option awards to eligible individuals who are foreign nationals or are individuals who are employed outside the State of Israel or the United States to recognize differences in local law, tax policy or custom, in order to effectuate the purposes of the 2024 Plan but without amending the 2024 Plan.
Eligibility. The 2024 Plan provides for granting awards under various tax regimes, including, without limitation, in compliance with Section 102 or Section 3(i) of the Israeli Tax Ordinance (the “Ordinance”), and for awards granted to our United States employees or service providers, including those who are deemed to be residents of the United States for tax purposes, Section 422 of the Code and Section 409A of the Code.
Grants. All awards granted pursuant to the 2024 Plan will be evidenced by an award agreement, in a form approved, from time to time, by the administrator in its sole discretion. The award agreement will set forth the terms and conditions of the award, including the type of award, number of shares subject to such award, vesting schedule and conditions (including performance goals or measures) and the exercise price, if applicable. Certain awards under the 2024 Plan may constitute or provide for a deferral of compensation, subject to Section 409A of the Code, which may impose additional requirements on the terms and conditions of such awards.
159
Unless otherwise determined by the administrator and stated in the award agreement, and subject to the conditions of the 2024 Plan, awards vest and become exercisable under the following schedule: 25% of the shares covered by the award on the first anniversary of the vesting commencement date determined by the administrator (and in the absence of such determination, the date on which such award was granted) and 6.25% of the shares covered by the award at the end of each subsequent three-month for a three year period; provided that the grantee remains continuously as an employee or provides services to the company throughout such vesting dates.
Each award will expire ten years from the date of the grant thereof, unless a shorter term of expiration is otherwise designated by the administrator.
Awards. The 2024 Plan provides for the grant of options (including incentive stock options and nonqualified stock options), Holdco Ordinary Shares, restricted shares, RSUs, stock appreciation rights and other share-based awards.
Options granted under the 2024 Plan to the Company employees who are U.S. residents may qualify as “incentive stock options” within the meaning of Section 422 of the Code, or may be non-qualified stock options. The exercise price of an option may not be less than the par value of the shares (if the shares bear a par value) for which such option is exercisable. The exercise price of an Incentive Stock Option may not be less than 100% of the fair market value of the underlying share on the date of grant or such other amount as may be required pursuant to the Code, and in the case of Incentive Stock Options granted to ten percent (10%) shareholders, not less than 110%.
Exercise. An award under the 2024 Plan may be exercised by providing the Company with a written or electronic notice of exercise and full payment of the exercise price for such shares underlying the award, if applicable, in such form and method as may be determined by the administrator and permitted by applicable law. An award may not be exercised for a fraction of a share. With regard to tax withholding, exercise price and purchase price obligations arising in connection with awards under the 2024 Plan, the administrator may, in its discretion, accept cash, provide for net withholding of shares in a cashless exercise mechanism or direct a securities broker to sell shares and deliver all or a part of the proceeds to the Company or the trustee.
Transferability. Other than by will, the laws of descent and distribution or as otherwise provided under the 2024 Plan, neither the options nor any right in connection with such options is assignable or transferable.
Termination of Employment. In the event of termination of a grantee’s employment or service with the Company or any of its affiliates, all vested and exercisable awards held by such grantee as of the date of termination may be exercised within three months after such date of termination, unless otherwise determined by the administrator, but in no event later than the date of expiration of the award as set forth in the award agreement. After such three-month period, all such unexercised awards will terminate and the shares covered by such awards become available for issuance under the 2024 Plan.
In the event of termination of a grantee’s employment or service with the Company or any of its affiliates due to such grantee’s death or permanent disability, or in the event of the grantee’s death within the one-year period (or such longer period as determined by the administrator) following his or her termination of service, all vested and exercisable awards held by such grantee as of the date of termination may be exercised by the grantee or the grantee’s legal guardian, estate or by a person who acquired the right to exercise the award by bequest or inheritance, as applicable, within one year after such date of termination, unless otherwise provided by the administrator, but in no event later than the date of expiration of the award as set forth in the award agreement. Any awards which are unvested as of the date of such termination or which are vested but not then exercised within the one-year period following such date will terminate and the shares covered by such awards shall again be available for issuance under the 2024 Plan.
Notwithstanding any of the foregoing, if a grantee’s employment or services with the Company or any of its affiliates is terminated for “cause” (as defined in the 2024 Plan), all outstanding awards held by such grantee (whether vested or unvested) will terminate on the date of such termination and the shares covered by such awards shall again be available for issuance under the 2024 Plan.
Voting Rights. Except with respect to restricted share awards, grantees will not have rights as a shareholder of the Company with respect to any shares covered by an award until the award has vested and the grantee has exercised such award, paid any exercise price for such award and becomes the record holder of the shares. With respect to restricted share awards, grantees will possess all incidents of ownership of the restricted shares, including the right to vote and receive dividends on such shares.
160
Dividends. Grantees holding restricted share awards will be entitled to receive dividends and other distributions with respect to the shares underlying the restricted share award. Any share subdivision, share dividend, consolidation of shares or similar transaction will be subject to the restrictions of the original restricted share award.
Transactions. In the event of a share split, reverse share split, share subdivision, recapitalization, consolidation or reclassification of the Company’s shares, a merger, consolidation, amalgamation or like transaction the administrator in its sole discretion may, and where required by applicable law shall, without the need for a consent of any holder, make an appropriate adjustment in order to adjust (i) the number and class of shares reserved and available for the outstanding awards, (ii) the number and class of shares covered by outstanding awards, (iii) the exercise price per share covered by any award, (iv) the terms and conditions concerning vesting and exercisability and the term and duration of the outstanding awards, (v) the type or class of security, asset or right underlying the award (which need not be only that of the Company, and may be that of the surviving corporation or any affiliate thereof or such other entity party to any of the above transactions), and (vi) any other terms of the award that in the opinion of the administrator should be adjusted; provided that any fractional shares resulting from such adjustment shall be rounded to the nearest whole share unless otherwise determined by the administrator. In the event of a distribution of a cash dividend to all shareholders, the administrator may determine, without the consent of any holder of an award, that the exercise price of an outstanding and unexercised award shall be reduced by an amount equal to the per share gross dividend amount distributed by the Company, subject to applicable law.
In the event of a merger or consolidation of the Company or a sale of all, or substantially all, of the Company’s shares or assets or other transaction having a similar effect on the Company, or change in the composition of the board of directors, or liquidation or dissolution, or such other transaction or circumstances that our board of directors determines to be a relevant transaction, then without the consent of the grantee, (i) unless otherwise determined by the administrator, any outstanding award will be assumed or substituted by such successor corporation, and (ii) regardless of whether or not the successor corporation assumes or substitutes the award, the administrator will (a) provide the grantee with the option to exercise the award as to all or part of the shares, and may provide for an acceleration of vesting of unvested awards, (b) cancel the award and pay in cash, shares of the Company, the acquirer or other corporation which is a party to such transaction or other property as determined by the administrator as fair in the circumstances, or (c) provide that the terms of any award shall be otherwise amended, modified or terminated, as determined by the administrator to be fair in the circumstances.
2024 Employee Share Purchase Plan
Immediately prior to the completion of the Business Combination, we adopted the 2024 Employee Share Purchase Plan (the “ESPP”). The ESPP is comprised of two distinct components: (1) the component intended to qualify for favorable U.S. federal tax treatment under Section 423 of the Code (the “Section 423 Component”) and (2) the component not intended to be tax qualified under Section 423 of the Code to facilitate participation for employees who are not eligible to benefit from favorable U.S. federal tax treatment and, to the extent applicable, to provide flexibility to comply with non-U.S. law and other considerations (the “Non-Section 423 Component”).
Number of Shares. A total of 1,646,604 Ordinary Shares (representing 5% of the Holdco Ordinary Shares outstanding immediately following the Closing) (the “Initial ESPP Share Pool”) are available for sale under the ESPP, subject to adjustment as provided for in the ESPP. In addition, on the first day of each calendar year beginning on January 1, 2025 and ending on and including January 1, 2032, such pool of Ordinary Shares shall be increased by that number of our Ordinary Shares equal to the lesser of: (i) 0.5% of the shares outstanding as of the last day of the immediately preceding calendar year, as determined on a fully diluted basis; or (ii) such smaller number as our board of directors may determine, if so determined prior to the January 1st of the calendar year in which the increase will occur, in each case as may be adjusted pursuant to the ESPP.
In no event will more than the Initial ESPP Share Pool be available for issuance under the Section 423 Component.
ESPP Administration. Unless otherwise determined by our board of directors, the compensation committee of our board of directors will administer the ESPP and will have the authority to interpret the terms of the ESPP and determine eligibility under the ESPP, to impose a mandatory holding period under which employees may not dispose or transfer shares under the ESPP, prescribe, revoke and amend forms, rules and procedures relating to the ESPP, to amend, suspend or terminate the ESPP and otherwise exercise such powers and to perform such acts as the administrator deems necessary or expedient to promote the best interests of the Company and its subsidiaries and to carry out the intent that the ESPP be treated as an “employee stock purchase plan” within the meaning of Section 423 of the Code for the Section 423 Component. The Administrator may adopt sub-plans applicable to designated subsidiaries or locations, which sub-plans may be designed to be outside the scope of Section 423 of the Code.
161
Eligibility. Participation in the Section 423 Component may be limited in the terms of any offering to employees of the Company and any of its designated subsidiaries (a) who customarily work 20 hours or more per week, (b) whose customary employment is for more than five months per calendar year and (c) who satisfy the procedural enrollment and other requirements set forth in the ESPP. Under the Section 423 Component, designated subsidiaries include any subsidiary (within the meaning of Section 424(f) of the Code) of the Company that has been designated by our board of directors or the compensation committee as eligible to participate in the ESPP (and if an entity does not so qualify within the meaning of Section 424(f) of the Code, it shall automatically be deemed to be a designated subsidiary in the Non-Section 423 Component). In addition, with respect to the Non-Section 423 Component, designated subsidiaries may include any corporate or noncorporate entity in which the Company has a direct or indirect equity interest or significant business relationship. Under the Section 423 Component, no employee may be granted a purchase right if, immediately after the purchase right is granted, the employee would own (or, under applicable statutory attribution rules, would be deemed to own) shares possessing 5% or more of the total combined voting power or value of all classes of shares of the Company or any of its subsidiaries. In addition, in order to facilitate participation in the ESPP, the compensation committee may provide for such special terms applicable to participants who are citizens or residents of a non-U.S. jurisdiction, or who are employed by a designated subsidiary outside of the U.S., as the compensation committee may consider necessary or appropriate to accommodate differences in local law, tax policy or custom. Except as permitted by Section 423 of the Code, with respect to the Section 423 Component, such special terms may not be more favorable than the terms of rights granted under the Section 423 Component to eligible employees who are residents of the United States.
Offering Periods. The ESPP provides for offering periods, not to exceed 27 months each, during which we will grant rights to purchase our Ordinary Shares to our eligible employees. The timing of the offering periods will be determined by the administrator. The terms and conditions applicable to each offering period will be set forth in an offering document adopted by the administrator for the particular offering period. The provisions of offerings during separate offering periods under the ESPP need not be identical.
Contributions. The ESPP will permit participants to purchase our Ordinary Shares through contributions (in the form of payroll deductions, or otherwise, to the extent permitted by the administrator). The percentage of compensation designated by an eligible employee as payroll deductions for participation in an offering may not be less than 1% and may not be more than the maximum percentage specified by the administrator in the applicable offering document (which maximum percentage shall be 20% in the absence of any such specification). A participant may increase or decrease the percentage of compensation designated in his or her subscription agreement, or may suspend his or her payroll deductions, at any time during an offering period; provided, however, that the administrator may limit the number of changes a participant may make in the applicable offering document. In the absence of any specific designation by the administrator, a participant may decrease (but not increase) his or her payroll deduction elections one time during each offering period.
Exercise of Purchase Right. Amounts contributed and accumulated by the participant will be used to purchase our Ordinary Shares at the end of each offering period. Unless otherwise determined by the administrator, the purchase price of the shares will be 85% of the lower of the fair market value of our Ordinary Shares on (i) the first trading day of the offering period or (ii) the last trading day of the offering period (and may not be lower than such amount with respect to the Section 423 Component). Participants may end their participation at any time during an offering period and will be paid their accrued contributions that have not yet been used to purchase our Ordinary Shares. Participation ends automatically upon termination of employment with us.
Non-Transferability. A participant may not transfer contributions credited to his or her account nor any rights granted under the ESPP other than by will, the laws of descent and distribution or as otherwise provided under the ESPP.
Corporate Transactions. In the event of certain transactions or events such as a consolidation, merger or similar transaction, a sale or transfer of all or substantially all of the Company’s assets, or a dissolution or liquidation of the Company, the administrator may, in its discretion, provide that (i) each outstanding purchase right will be (a) assumed or substituted for a right granted by the acquiror or successor corporation or by a parent or subsidiary of such entity, (b) terminated in exchange for cash or other property as determined by the administrator or (c) cancelled with accumulated payroll deductions returned to each participant, or (ii) the participant’s accumulated payroll deductions may be used to purchase shares prior to the end of the offering period and before the date of the proposed sale, merger or similar transaction.
Amendment; Termination. The administrator will have the authority to amend, suspend or terminate the ESPP, subject to the exceptions set forth in the ESPP. The ESPP is not subject to a specific termination date.
162
| C. | Board Practices |
Board of Directors
Under the Companies Law and the Amended Articles, Holdco’s business and affairs are managed under the direction of our board of directors. Our board of directors may exercise all powers and may take all actions that are not specifically granted to our shareholders or to executive management. Our Chief Executive Officer (referred to as a “general manager” under the Companies Law) is responsible for our day-to-day management. Our Chief Executive Officer is appointed by, and serves at the discretion of, our board of directors, subject to the employment agreement that we have entered into with her. All other executive officers are appointed by the Chief Executive Officer, subject to applicable corporate approvals, and are subject to the terms of any applicable employment or consulting agreements that we may enter into with them.
Under the Amended Articles, our board of directors must consist of not less than three but no more than 11 directors divided into three classes with staggered three-year terms. Each class of directors consists, as nearly as possible, of one-third of the total number of directors constituting the entire board of directors. At each annual general meeting of our shareholders, the election or re-election of directors following the expiration of the term of office of the directors of that class of directors will be for a term of office that expires on the third annual general meeting following such election or re-election, such that from our first annual general meeting and thereafter, each year the term of office of only one class of directors will expire.
Our directors are divided among three classes as follows:
| ● | the Class I director is Laurence Klein, and his term will expire at the annual general meeting of shareholders following the closing of the Business Combination; |
| ● | the Class II directors are Gerald Ostrov and Robert Powell, and their terms will expire at the annual general meeting of shareholders held after the annual general meeting referred to in the bullet point immediately above; and |
| ● | the Class III directors are Christina Spade and Adriana Machado, and their terms will expire at the annual general meeting of shareholders held after the annual general meeting referred to in the bullet point immediately above. |
Pursuant to the Amended Articles, our directors will be appointed by a simple majority vote of holders of Holdco Ordinary Shares and Holdco Preferred Shares voting as a single class, participating and voting at an annual general meeting of our shareholders, provided that (i) in the event of a contested election, the method of calculation of the votes and the manner in which the resolutions will be presented to our shareholders at the general meeting will be determined by our board of directors in its discretion, and (ii) in the event that our board of directors does not or is unable to make a determination on such matter, then the directors will be elected by a plurality of the voting power represented at the general meeting in person or by proxy and voting on the election of directors.
Each director will hold office until the annual general meeting of our shareholders for the year in which such director’s term expires, unless the tenure of such director expires earlier pursuant to the Companies Law or unless such director is removed from office as described below.
Under the Amended Articles, the approval of the holders of at least 65% of the total voting power of our shareholders will generally be required to remove any of our directors (other than any external director, if appointed) from office. Any amendment to this provision shall require the approval of at least 65% of the total voting power of our shareholders. In addition, vacancies on our board of directors may only be filled by a vote of a simple majority of the directors then in office (or, if the board of directors determines, the general meeting). A director so appointed will hold office until the next annual general meeting of our shareholders for the election of the class of directors in respect of which the vacancy was created, or in the case of a vacancy due to the number of directors being less than the maximum number of directors stated in the Amended Articles, the new director filling the vacancy will serve until the next annual general meeting of our shareholders for the election of the class of directors to which such director was assigned by our board of directors.
163
Chairperson of the Board of Directors
The Amended Articles provide that the chairperson of our board of directors is appointed by the members of our board of directors from among them. Under the Companies Law, the chief executive officer of a public company, or a relative of the chief executive officer, may not serve as the chairperson of the board of directors of such public company, and the chairperson of the board of directors of a public company, or a relative of the chairperson, may not be vested with authorities of the chief executive officer of such public company without shareholder approval consisting of a majority vote of the shares present and voting at a shareholders meeting and approval of the audit committee, and in addition, either:
| ● | at least a majority of the shares of non-controlling shareholders and shareholders that do not have a personal interest in the approval voted at the meeting are voted in favor (disregarding abstentions); or |
| ● | the total number of shares of non-controlling shareholders and shareholders who do not have a personal interest in such appointment that voted against such appointment does not exceed two percent (2%) of the aggregate voting rights in the company. |
The shareholders’ approval can be effective for a period of up to five years following an initial public offering, and subsequently, for additional periods of up to three years.
In addition, a person who is subordinated, directly or indirectly, to the chief executive officer may not serve as the chairperson of the board of directors; the chairperson of the board of directors may not be vested with authorities that are granted to persons who are subordinated to the chief executive officer; and the chairperson of the board of directors may not serve in any other position in the company or in a controlled subsidiary, but may serve as a director or chairperson of a controlled subsidiary.
External Directors
Under the Companies Law, companies incorporated under the laws of the State of Israel that are “public companies,” including companies with shares listed on Nasdaq, are required to appoint at least two external directors. Pursuant to regulations promulgated under the Companies Law, companies with shares traded on certain U.S. stock exchanges, including Nasdaq, which do not have a “controlling shareholder,” as defined in the Companies Law, and which comply with the independent director requirements and the audit committee and compensation committee composition requirements of U.S. federal securities law and the U.S. stock exchange applicable to domestic issuers, may, subject to certain other conditions, “opt out” from the Companies Law requirements to appoint external directors and related Companies Law rules concerning the composition of the audit committee and compensation committee of the board of directors. In accordance with these regulations, we have determined that we currently qualify for such exemption, and have consequently elected to “opt out” from the Companies Law requirement to appoint external directors and related Companies Law rules concerning the composition of the audit committee and compensation committee of our board of directors. However, we will review this determination from time to time and if we find that we have ceased to qualify for such exemption we will need to comply with the applicable rules of the Companies Law relating to the appointment of external directors, their inclusion in our audit committee and compensation committee and their presence for establishing a quorum in such committees, among other matters.
Director Independence
Nasdaq listing standards require that a majority of our board of directors be independent. An “independent director” is defined generally as a person who has no material relationship with the listed company (either directly or as a partner, stockholder, shareholder or officer of an organization that has a relationship with the listed company). Our board of directors has determined that Gerald Ostrov, Christina Spade and Adriana Machado are “independent directors” as defined in Nasdaq listing standards and applicable SEC rules.
164
Committee of the Board of Directors
Audit Committee
Companies Law Requirements
Under the Companies Law, the board of directors of a public company must appoint an audit committee. The audit committee must be composed of at least three directors.
Listing Requirements
Under the corporate governance rules of Nasdaq, we are required to maintain an audit committee consisting of at least three independent directors, each of whom is financially literate and one of whom has accounting or related financial management expertise.
Our audit committee consists of Christina Spade, Adriana Machado and Gerald Ostrov, and Christina Spade serves as the chairperson of the audit committee. All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and the corporate governance rules of Nasdaq. Our board of directors has determined that Christina Spade is an audit committee financial expert as defined by the SEC rules and has the requisite financial experience as defined by the corporate governance rules of Nasdaq.
Our board of directors has determined that each member of our audit committee is “independent” as such term is defined in Rule 10A-3(b)(1) under the Exchange Act, which is different from the general test for independence of board and committee members.
Audit Committee Role
Our board of directors has adopted an audit committee charter that sets forth the responsibilities of the audit committee consistent with the Companies Law, the SEC rules and the corporate governance rules of Nasdaq, which includes:
| ● | retaining and terminating our independent auditors, subject to ratification by our board of directors, and in the case of retention, to ratification by the shareholders; |
| ● | pre-approving audit and non-audit services to be provided by the independent auditors and related fees and terms; |
| ● | overseeing the accounting and financial reporting processes of our company and audits of our financial statements, the effectiveness of our internal control over financial reporting and making such reports as may be required of an audit committee under the rules and regulations promulgated under the Exchange Act; |
| ● | reviewing with management and our independent auditor our annual and quarterly financial statements prior to publication or filing (or submission, as the case may be) to the SEC; |
| ● | recommending to our board of directors the retention and termination of the internal auditor, and the internal auditor’s engagement fees and terms, in accordance with the Companies Law as well as approving the yearly or periodic work plan proposed by the internal auditor; |
| ● | reviewing with our general counsel and/or external counsel, as deemed necessary, legal and regulatory matters that could have a material impact on the financial statements; |
| ● | identifying irregularities in our business administration, inter alia, by consulting with the internal auditor or with the independent auditor, and suggesting corrective measures to our board of directors; |
| ● | reviewing policies and procedures with respect to transactions (other than transactions related to the compensation or terms of services) between us and our officers and directors, or affiliates of our officers or directors, or transactions that are not in the ordinary course of our business and deciding whether to approve such acts and transactions if required under the Companies Law; and |
| ● | establishing procedures for the handling of employees’ complaints as to the management of our business and the protection to be provided to such employees. |
165
Compensation Committee
Companies Law Requirements
Under the Companies Law, the board of directors of a public company must appoint a compensation committee, which must be composed of at least three directors.
Listing Requirements
Under the corporate governance rules of Nasdaq, we are required to maintain a compensation committee consisting of at least two independent directors.
Our compensation committee consists of Gerald Ostrov and Christina Spade, and Gerald Ostrov serves as chairperson of the compensation committee. Our board of directors has determined that each member of our compensation committee is independent under the corporate governance rules of Nasdaq, including the additional independence requirements applicable to the members of a compensation committee.
Compensation Committee Role
In accordance with the Companies Law, the roles of the compensation committee are, among others, as follows:
| ● | making recommendations to our board of directors with respect to the approval of the compensation policy for office holders and, once every three years, regarding any extensions to a compensation policy that was adopted for a period of more than three years; |
| ● | reviewing the implementation of the compensation policy and periodically making recommendations to our board of directors with respect to any amendments or updates of the compensation policy; |
| ● | resolving whether or not to approve arrangements with respect to the terms of office and employment of office holders; and |
| ● | exempting, under certain circumstances, a transaction with our Chief Executive Officer from the approval of our shareholders. |
An “office holder” is defined in the Companies Law as a general manager, chief business manager, deputy general manager, vice general manager, any other person assuming the responsibilities of any of these positions regardless of such person’s title, a director and any other manager directly subordinate to the general manager. Certain of the persons listed in the table under the section titled "Executive Officers and Directors” are office holders under the Companies Law.
Our board of directors adopted a compensation committee charter that sets forth the responsibilities of the committee consistent with the corporate governance rules of Nasdaq and the Companies Law, which includes:
| ● | recommending to our board of directors for its approval a compensation policy in accordance with the requirements of the Companies Law, as well as other compensation policies, incentive-based compensation plans and equity-based compensation plans, and overseeing the development and implementation of such policies and recommending to our board of directors any amendments or modifications the committee deems appropriate, including as required under the Companies Law; |
| ● | reviewing and approving the granting of options and other incentive awards to our Chief Executive Officer and other executive officers, including reviewing and approving corporate goals and objectives relevant to the compensation of our Chief Executive Officer and other executive officers, including evaluating their performance in light of such goals and objectives; |
| ● | approving and exempting certain transactions regarding office holders’ compensation pursuant to the Companies Law; and |
| ● | administering our equity-based compensation plans, including without limitation, approving the adoption of such plans, amending and interpreting such plans and the awards and agreements issued pursuant thereto, and making awards to eligible persons under the plans and determining the terms of such awards. |
166
Nominating, Governance and Sustainability Committee
The Companies Law does not require us to have a nominating committee. Nevertheless, our board of directors has decided to form a nominating, governance and sustainability committee which consists of Adriana Machado and Gerald Ostrov, and Adriana Machado serves as chairman of the nominating, governance and sustainability committee. Our board of directors has adopted a nominating, governance and sustainability committee charter that sets forth the responsibilities of the committee, which includes:
| ● | overseeing and assisting our board in reviewing and recommending nominees for election as directors; |
| ● | assessing the performance of the members of our board; |
| ● | establishing and maintaining effective corporate governance policies and practices, including, but not limited to, developing and recommending to our board a set of corporate governance guidelines applicable to our business; and |
| ● | overseeing our policies, programs and strategies related to ESG. |
| D. | Employees |
Following and as a result of the Business Combination, the business of the Company is conducted through Nuvo, its direct, wholly-owned subsidiary.
As of December 31, 2023, we had 33 full-time and 3 part-time employees in Israel, 14 full-time contractors in Ukraine and 9 full-time employees in the United States. Our operations in Israel have not been materially affected by the war between Israel and Hamas, however, see “Risk Factors — Risks Related to Israeli Law and Our Operations in Israel — Conditions in Israel, including the ongoing war between Israel and Hamas, and other conflicts in the region, may adversely affect our business, our results of operations and our ability to raise additional funds.” Following the invasion of Ukraine by Russia in 2022, we closed our Ukraine office and all of our Ukrainian employees work remotely. We believe that the success of our business will depend, in part, on our ability to attract and retain qualified personnel. Our human capital strategy is closely aligned with our vision and focuses on attracting, retaining, developing and engaging top talent. We monitor our success with insights across human capital metrics such as hire per plan, professional growth and promotions, performance and employee development feedback, and turnover. None of our employees are represented by a labor union or are a party to a collective bargaining agreement, and we believe that we have good relations with our employees.
| E. | Share Ownership |
Information about the ownership of Holdco Ordinary Shares by our directors and members of senior management is set forth in Item 7.A of this Annual Report. Information about arrangements for involving employees in the capital of the Company is set forth in Item 6.B of this Annual Report.
| F. | Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation |
None.
167
| ITEM 7. | MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS |
| A. | Major Shareholders |
The following table sets forth information regarding the beneficial ownership of Holdco Ordinary Shares as of May 1, 2024 immediately following the consummation of the Business Combination by:
| ● | each person known by us to be the beneficial owner of more than 5% of the Holdco Ordinary Shares; |
| ● | each of our directors and our executive officers; and |
| ● | all our directors and executive officers. |
Except as otherwise noted herein, the number and percentage of Holdco Ordinary Shares beneficially owned is determined in accordance with Rule 13d-3 of the Exchange Act, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rule, beneficial ownership includes any Holdco Ordinary Shares as to which the holder has sole or shared voting power or investment power and also any Holdco Ordinary Shares which the holder has the right to acquire within 60 days of the Closing Date through the exercise of any option, warrant or any other right.
We have based percentage ownership on 33,261,549 Holdco Ordinary Shares outstanding as of the Closing Date, May 1, 2024.
| Number | Percentage | |||||||
| Name and Address of Beneficial Owner | ||||||||
| Directors and Executive Officers of Holdco:** | ||||||||
| Robert Powell | 28,566 | * | ||||||
| Douglas Blankenship | 385 | * | ||||||
| Laurence Klein(2)(3)(4) | 3,053,709 | 9.2 | % | |||||
| Christina Spade | 20,000 | * | ||||||
| Gerald Ostrov(5) | 485,847 | 1.5 | % | |||||
| Adriana Machado | 20,000 | * | ||||||
| All directors and executive officers as a group (six individuals) | 3,608,507 | 10.8 | % | |||||
| 5% or More Holders:*** | ||||||||
| LAMF SPAC Holdings I LLC(1) | 7,311,372 | (6) | 21.9 | % | ||||
| Axxion SA(acting on behalf of the German UCITS Funds “Frankfurter Aktienfonds für Stiftungen”) | 2,337,328 | 7.0 | % | |||||
| Laurence Klein | 3,053,709 | 9.2 | % | |||||
| * | Less than one percent. |
| ** | Other than with respect to Christina Spade and Adriana Machado, the beneficial ownership information of the directors and executive officers of Holdco is based on Nuvo Shares beneficially owned by such persons, as of the date of closing, May 1, 2024. |
| *** | Other than with respect to LAMF SPAC Holdings I LLC or as otherwise noted below, the beneficial ownership information of the 5% of more holders of Holdco is based on Nuvo Shares beneficially owned by such persons, as of the date of closing, May 1, 2024. |
168
| (1) | LAMF SPAC Holdings I LLC is the record holder of the shares reported herein. LAMF SPAC I LLC is the managing member of LAMF SPAC Holdings I LLC. LAMF SPAC I LLC has voting and investment discretion with respect to the ordinary shares held of record by LAMF SPAC Holdings I LLC. There are three managing members of LAMF SPAC I LLC. Each managing member has one vote, and the approval of a majority is required to approve an action. Under the so-called “rule of three,” voting and dispositive decisions regarding an entity’s securities are made by three or more individuals, and voting or dispositive decisions require the approval of a majority of those individuals, then none of the individuals is deemed a beneficial owner of the entity’s securities. Based on the foregoing, no individual managing member of LAMF SPAC I LLC exercises voting or dispositive control over any of the shares held by the entity, even those in which he holds a pecuniary interest. Accordingly, none of them will be deemed to have or share beneficial ownership of such shares. Above reflects the ownership of LAMF SPAC Holdings I LLC prior to distribution of Holdco securities to certain of its members on or about May 1, 2024. |
| (2) | Consists of (i) 424,503 Nuvo Shares held directly by Nuvo Investors LLC and indirectly by Laurence Klein as Managing Director of Nuvo Investors LLC and (ii) 514,606 Nuvo Shares Nuvo Investors LLC received upon conversion of certain Nuvo SAFEs upon the consummation of the Business Combination. Mr. Klein exercises sole voting and investment power with respect to the securities held by Nuvo Investors LLC. The address for Nuvo Investors LLC is 803 Wildwood Road, West Hempstead, NY 11552. |
| (3) | Consists of 480,693 Nuvo Shares held directly by Nalay, Inc. and indirectly by Laurence Klein as President of Nalay, Inc. Mr. Klein exercises sole voting and investment power with respect to the securities held by Nalay, Inc. The address for Nalay, Inc. is 803 Wildwood Road, West Hempstead, NY 11552. |
| (4) | Consists of 9,167 Nuvo Shares held directly by LCK Holdings LLC and indirectly by Laurence Klein as Managing Director of LCK Holdings LLC. Mr. Klein exercises sole voting and investment power with respect to the securities held by LCK Holdings LLC. The address for LCK Holdings LLC is 803 Wildwood Road, West Hempstead, NY 11552. |
| (5) | Includes 16,566 Nuvo Shares issuable upon the exercise of options and 82,093 Nuvo Shares issuable upon the exercise of the Bridge Loan warrants within 60 days following May 1, 2024. |
| (6) |
In connection with the Extension, the Sponsor agreed to transfer to certain unaffiliated third party investors (i) for the initial extension of LAMF’s deadline to consummate an initial business combination, 606,480 LAMF Class A Ordinary Shares, and (ii) 101,080 Founder Shares for each additional monthly extension, or up to an aggregate of 1,212,960 LAMF Class A Ordinary Shares if the initial extension and all additional monthly extensions are implemented. |
169
| B. | Related Party Transactions |
Shareholder Support Agreement and Lockup
Concurrently with the execution of the Business Combination Agreement, LAMF, Nuvo, Holdco and certain Nuvo Shareholders entered into the Shareholder Support Agreement, pursuant to which such Nuvo Shareholders agreed, among other things, to vote any Nuvo Shares held by them in favor of the Business Combination, the Acquisition Merger, and such other actions as contemplated in the Business Combination Agreement for which the approval of the Nuvo Shareholders is required.
Pursuant to the Shareholder Support Agreement, the Nuvo Lockup Parties are subject to the Nuvo Lock-up, which generally restricts transfers on the Holdco Ordinary Shares (or any instruments exercisable or exchangeable for, or convertible into, Holdco Ordinary Shares) held by each such Nuvo Lock-up Party as of the Closing Date for the six month period following the Closing Date, subject to certain customary exceptions. With the exception the Nuvo Lockup Exempt Shareholders, who are those Nuvo Shareholders holding less than one percent of the Holdco Ordinary Shares outstanding immediately after Closing and are exempt pursuant the Amended Articles, all Nuvo Shareholders are subject to a six month restriction on transfers of Holdco Ordinary Shares and Holdco Preferred Shares effective as of the Closing pursuant to the Amended Articles, subject to exceptions as contained therein.
Registration Rights Agreement
At the Closing, LAMF, Nuvo, Holdco, Sponsor, Simon Horsman, Jeffrey Soros, Morgan Earnest, Christina Spade, Adriana Machado, and Michael Brown, as executive officers and/or directors of LAMF prior to the Closing, Keith Harris, as advisor to LAMF prior to the Closing, LAMF SPAC I LLC, Nweis Investments LLC, Atoe LLC, 10X LAMF SPC SPV LLC, 10X LLC, ASCJ Global LLC – Series 16, and Cohen Sponsor LLC – A16 RS, as the members of the Sponsor, certain Nuvo Shareholders, and the executive officers and directors of Nuvo prior to the Closing, entered into the Registration Rights Agreement, pursuant to which, among other things, Holdco agreed to register for resale, pursuant to Rule 415 under the Securities Act, the Registerable Securities (as defined in the Registration Rights Agreement) that are held by the parties thereto from time to time. The parties are entitled to certain customary demand and piggyback registration rights under the Registration Rights Agreement, which are subject to customary terms and conditions, including with respect to cooperation and reduction of underwritten shelf takedown provisions, with respect to the securities of Holdco.
Interim Financing Agreements
Prior to, upon and following the execution of the Business Combination Agreement, Nuvo and Holdco entered into securities purchase agreements (the “Interim Financing Agreements”) with certain investors (the “Interim Financing Investors”) pursuant to which (i) Nuvo issued Nuvo Crossover Preferred Shares to the Interim Financing Investors, which were exchanged for an aggregate of 1,850,126 Holdco Preferred Shares in connection with the Closing, and (ii) upon the Closing, Holdco issued an aggregate of 3,823,530 Holdco Ordinary Shares to the Interim Financing Investors, which shares are not registered under the Securities Act in connection with the Business Combination Agreement, and which provided Nuvo with an aggregate of approximately $13,000,000 of gross proceeds as a result of the Interim Financing. Certain of the Interim Financing Investors are affiliated with LAMF and the Sponsor and invested an aggregate of $2,000,000 in the Interim Financing (such investors the “Sponsor Investors”). These affiliates are: (i) Jeffrey Soros, LAMF’s Chairman, who invested $500,000, (ii) Tamim Mourad, a strategic investor of LAMF and an affiliate of a member of the Sponsor, who invested $500,000 and (iii) Gaingels 10X Capital Diversity Fund I, LP, a Delaware limited partnership and an affiliate of a member of the Sponsor, that invested $1,000,000.
The following summarizes a number of the key provisions in the Interim Financing Agreements:
| ● | Except for the Interim Financing Agreement with LAMF, Nuvo was precluded from creating, authorizing or issuing any shares with the same or more favorable rights as those associated with the shares purchased pursuant to the Interim Financing Agreements. |
| ● | Nuvo committed to entering into a registration rights agreement with the Interim Financing Investors within 60 days from the consummation of the Interim Financing to register the resale, pursuant to Rule 415 under the Securities Act, of certain Holdco Ordinary Shares and other equity securities of Holdco that are held by such parties thereto from time to time. |
| ● | Holdco committed to delivering (and Nuvo committed to causing Holdco to deliver) Interim Financing Incentive Shares immediately following the Closing. |
170
| ● | Nuvo committed to various reporting and information rights for the Interim Financing Investors, including annual audited financial statements and quarterly financial statements. |
The Interim Financing Agreements contain customary representations and warranties and an indemnity in favor of the Interim Financing Investors for breaches by Nuvo of its covenants, representations and warranties thereunder. The Interim Financing Agreement with LAMF includes several additional conditions precedent to LAMF’s participation in the Interim Financing, including without limitation, Nuvo’s execution of the Philips MPA as well as the Company and LAMF executing the BCA.
In connection with the Interim Financing Agreements, Nuvo obtained the requisite approvals and filed an amended & restated articles of association, which authorized the formation and issuance of the Nuvo Crossover Preferred Shares. Pursuant to Nuvo’s amended & restated articles of association, the Interim Financing Investors received a liquidation and dividend preference, ranking them ahead of all other classes of Nuvo shareholders, equal to the greater of (i) the sum of three times the original issuance price for the Nuvo Crossover Preferred Shares, or (ii) the amount such shareholders would actually receive if such Nuvo Crossover Preferred Shares had been converted into Nuvo Shares immediately prior to a distribution event; in each case, plus any dividends declared but unpaid on such share.
Nuvo SAFE Amendment
From June 2020 through April 2023, Nuvo issued to certain investors the Nuvo SAFEs, pursuant to which such investors invested cash in Nuvo in the aggregate principal amount of $22.97 million. For additional information on the original terms of the Nuvo SAFEs see, Item 5.B. “Operating and Financial Review and Prospects—Liquidity and Capital Resources—Sources of Liquidity”.
In August and September 2023, Nuvo obtained the necessary consents for the Nuvo SAFE Amendment, which was entered into in order to equalize the economic conversion terms across these series the Nuvo SAFEs, such that: (a) upon the consummation of an applicable equity financing, the purchase amount under each Nuvo SAFE shall convert into Nuvo’s shares at a 25% discount over the price per share in such financing round, provided that the pre-money valuation upon which the principal amount will convert into Nuvo shares will not exceed $200 million; and (b) immediately prior to the consummation of the Business Combination (which did not otherwise constitute a Liquidity Event pursuant to the Nuvo SAFE Amendment), the purchase amount under the Nuvo SAFEs converted into Nuvo Shares based on (1) a price per share determined by dividing $150 million by the then issued and outstanding share capital of Nuvo (assuming exercise or conversion of all outstanding vested and unvested options and/or warrants, convertible debt instruments and similar instruments, but excluding any SAFEs and other convertible instruments that are outstanding as of immediately prior to the Closing) and including all shares reserved and available for future grant under any equity incentive or similar plan of Nuvo, in each case as of immediately prior to the Closing, or (2) by the price per share imputed to the Nuvo Shares pursuant to the Business Combination Agreement multiplied by 75%, whichever resulted in the issuance to the Nuvo SAFE holder of a greater number of Nuvo Shares. Accordingly, upon and in connection with the Closing, Nuvo issued approximately 3.56 million Nuvo Shares in satisfaction and discharge of its obligations under the outstanding amended Nuvo SAFEs, which were exchanged for Holdco Ordinary Shares in connection with the Closing. The conversion of the Nuvo Convertible Loans (discussed below) and Nuvo SAFEs into Holdco Ordinary Shares in connection with the Closing result in the issuance of 5.24 million Holdco Ordinary Shares.
Nuvo Loan Amendment
During 2022 and 2023, Nuvo entered into certain loan agreements pursuant to which it borrowed from a number of lenders an aggregate principal amount of $7.9 million Nuvo Convertible Loans, of which $6.8 million in principal remains outstanding. For additional information on the original terms of the Nuvo Convertible Loans see, Item 5.B. “Operating and Financial Review and Prospects—Liquidity and Capital Resources—Sources of Liquidity”. In August and September 2023 Nuvo obtained the necessary consents for the Nuvo Loan Amendment, such that, in exchange for the Extension Fee under the original loan terms, the maturity date of each loan was extended to the earlier of the second anniversary of the applicable loan or the Closing. In addition, pursuant to the Nuvo Loan Amendment, each lender has agreed to apply the principal amount of the Nuvo Convertible Loan, the accrued and unpaid interest thereon and the Extension Fee to the purchase amount of the related Nuvo SAFE. See “ — Nuvo SAFE Amendment” above. As such, in connection with the Closing, Nuvo’s obligations under the Nuvo Convertible Loans converted to an aggregate SAFE purchase amount of approximately $12.25 million, which then converted, pursuant to the terms of the amended Nuvo SAFEs and Nuvo SAFE Amendment, into approximately 1.89 million Nuvo Shares which were then exchanged for Holdco Shares pursuant to the terms of the Business Combination Agreement.
171
Oren Oz Letter Agreement
On May 29, 2023, we entered into an amended & restated letter agreement (the “Letter Agreement”) with Oren Oz, our founder and one of our directors. Pursuant to the Letter Agreement, we issued to Mr. Oz options to purchase 346,575 Nuvo Ordinary Shares at an exercise price of NIS 0.01, which options were fully vested upon the date of grant, representing 333,194 Holdco Ordinary Shares following the Closing. In exchange for the issuance of the options, Mr. Oz agreed to the adoption by Nuvo of certain amended and restated articles of association, with such articles becoming effective upon the approval of our shareholders. In addition, we agreed to reimburse Mr. Oz for up to $50,000 plus VAT for expenses related to a tax ruling Mr. Oz may seek in connection with the tax treatment of such options. The Letter Agreement also provides for the payment to Mr. Oz of a severance payment in the amount of NIS 623,289 in respect of Mr. Oz’s former employment with Nuvo, with such severance payment to be paid no later than December 31, 2023. This payment has been extended and is currently being paid in monthly installments over a 5 ½ month period ending in August 2024. Pursuant to the Letter Agreement, by no later than January 31, 2024, Mr. Oz will receive the remaining portion of a termination payment in nine monthly payments, for an aggregate amount equal to NIS 1,026,603. These payments have been completed. In addition, we agreed to pay Mr. Oz amounts that were not yet distributed to his pension fund during the term of his employment, which such amounts are approximately NIS 110,978. We expect to pay this last obligation in the third quarter of 2024.
Consulting Agreement
On July 1, 2023, we entered into a consulting services agreement (the “Consulting Agreement”) with Adama GmbH, a German limited liability company (“Adama”), an entity wholly owned and controlled by Daniel Gilcher, a former director from April 2018 to July 2023 and our Interim Chief Financial Officer as of August 2023, pursuant to which Adama is to provide the consulting services set forth in the statement of work through its designated service provider, Mr. Gilcher. Pursuant to the Consulting Agreement, we agreed to pay Adama an annual retainer of $300,000 to be paid as follows: (i) 50% in cash and (ii) 50% by way of fully-vested options to purchase Nuvo Shares issued under the Nuvo 2015 Plan. The Consulting Agreement’s term expires June 30, 2024 and will be automatically renewed for one year terms unless mutually agreed otherwise and unless terminated in accordance with the Consulting Agreement. Pursuant to addendums entered into on September 10, 2023 and September 18, 2023, Mr. Gilcher served as Nuvo’s Interim Chief Financial Officer until Mr. Blankenship was onboarded as Nuvo’s full-time chief financial officer.
Bridge Financing
Since November 2023 Nuvo has been engaged in the Bridge Financing. Gaingels 10x Capital Diversity Fund I, LP is a Bridge Financing Holder, holding an aggregate principal amount of $1.0 million of Bridge Financing Notes and related warrants to purchase 273,647 Holdco Ordinary Shares, is an affiliate of a member of the Sponsor and serves as collateral agent with respect to the collateral securing the Bridge Financing Notes. Gerald Ostrov, a member of the Holdco Board, holds an aggregate principal amount of $636,000 of Bridge Financing Notes and related warrants to purchase 174,039 Holdco Ordinary Shares. Robert Powell, our Chief Executive Officer and a member of the Holdco Board, holds an aggregate principal amount of $100,000 of Bridge Financing Notes and related warrants to purchase 27,365 Holdco Ordinary Shares. For a description of the material terms of the Bridge Financing Notes see “Explanatory Note—Bridge Financing” above.
| C. | Interests of Experts and Counsel |
Not applicable.
172
| ITEM 8. | FINANCIAL INFORMATION |
| A. | Consolidated Statements and Other Financial Information |
Financial Statements
See Item 18 “Financial Statements.”
Legal Proceedings
We are not currently subject to any material legal proceedings although we may be subject to legal proceedings and claims in the ordinary course of business in the future. We cannot predict the results of any such disputes, and despite the potential outcomes, the existence thereof may have an adverse material impact on us due to diversion of management time and attention as well as the financial costs related to resolving such disputes.
Dividend Policy
We have never declared nor paid any dividends on the Holdco Ordinary Shares or Holdco Preferred Shares. We currently intend to retain future earnings, if any, to finance operations and expand our business. We do not anticipate paying any dividends in the foreseeable future. Our board of directors may declare a dividend to be paid to the holders of the Holdco Ordinary Shares or Holdco Preferred Shares, the form, frequency and amount which will depend upon our future operations and earnings, capital requirements, and surplus, general financial condition, contractual restrictions and other factors that our Board of Directors may deem relevant. Under the Companies Law, dividend distributions are determined by the board of directors and do not require the approval of the shareholders of a company unless the company’s articles of association provide otherwise. Our Amended Articles do not require shareholder approval of a dividend distribution and provide that dividend distributions may be determined by our board of directors.
Pursuant to the Companies Law, the distribution amount is limited to the greater of retained earnings or earnings generated over the previous two years, according to our then last reviewed or audited financial statements (less the amount of previously distributed dividends, if not reduced from the earnings), provided that the end of the period to which the financial statements relate is not more than six months prior to the date of the distribution. If we do not meet such criteria, then we may distribute dividends only with court approval. In each case, we are only permitted to distribute a dividend if our board of directors and, if applicable, the court determines that there is no reasonable concern that payment of the dividend will prevent us from satisfying our existing and foreseeable obligations as they become due.
In the event of our liquidation, after satisfaction of liabilities to creditors, and also in the event of a distribution to shareholders, our assets will be distributed first to the holders of the Holdco Preferred Shares in an amount equal to the greater of (i) the sum of three times the price originally paid for each Holdco Preferred Share, which as of the date hereof would result in an aggregate amount of $36,000,000 or (ii) the amount such holder would actually receive if such Holdco Preferred Share had been converted into Holdco Ordinary Shares immediately prior to the liquidation or distribution. Thereupon, the distributable assets will be distributed to the holders of the Holdco Ordinary Shareholders on a pari passu basis. This right, as well as the right to receive dividends, may be affected by the grant of preferential dividend or distribution rights to the holders of a class of shares with preferential rights that may be authorized in the future.
| B. | Significant Changes |
Not Applicable.
173
| ITEM 9. | THE OFFER AND LISTING |
| A. | Offer and Listing Details |
Not applicable.
| B. | Plan of Distribution |
Not applicable.
| C. | Markets |
The Holdco Ordinary Shares are listed on The Nasdaq Global Market under the symbol “NUVO” and the Holdco Warrants are listed on The Nasdaq Capital Market under the symbol “NUVOW”.
| D. | Selling Shareholders |
Not applicable.
| E. | Dilution |
Not applicable.
| F. | Expenses of the Issue |
Not applicable.
174
| ITEM 10. |
ADDITIONAL INFORMATION |
| A. | Share Capital |
Not applicable.
| B. | Articles of Association |
A copy of our Amended Articles is attached as Exhibit 1.1 to this Annual Report. The information called for by this Item is set forth in Exhibit 2.1 to this Annual Report and is incorporated by reference herein.
| C. | Material Contracts |
For a description of our material contracts, see Item 4.B. “Business Overview––Expanded Commercial Partnerships” and “––Commercial Relationships” for a description of the agreements related to our directors and officers, Item 6.B. “Compensation”.
| D. | Exchange Controls |
There are currently no Israeli currency control restrictions on payments of dividends or other distributions with respect to the Holdco Ordinary Shares or the proceeds from the sale of the Holdco Ordinary Shares, except for the obligation of Israeli residents to file reports with the Bank of Israel regarding certain transactions. However, legislation remains in effect pursuant to which currency controls can be imposed by administrative action at any time.
Non-residents of Israel who purchase our securities with non-Israeli currency will be able to repatriate dividends (if any), liquidation distributions and the proceeds of any sale of such securities, into non-Israeli currencies at the rate of exchange prevailing at the time of repatriation, provided that any applicable Israeli taxes have been paid (or withheld) on such amounts.
Neither our Amended Articles nor the laws of the State of Israel restrict in any way the ownership or voting of our ordinary shares by non-residents of Israel, except with respect to citizens of countries that are in a state of war with Israel.
| E. | Taxation |
Material U.S Federal Income Tax Considerations to U.S. Holders
The following discussion is a summary of the material U.S. federal income tax considerations applicable to you if you are a U.S. Holder (as defined below) as a consequence of the ownership and disposition of Holdco Ordinary Shares and Holdco Warrants. This discussion addresses only those U.S. Holders that hold Holdco Ordinary Shares and/or Holdco Warrants as capital assets within the meaning of Section 1221 of the Code (generally property held for investment).
175
This discussion does not address all U.S. federal income tax considerations that may be relevant to any particular investor’s particular circumstances, including the alternative minimum tax, the Medicare tax on certain investment income and the different consequences that may apply to investors subject to special rules under U.S. federal income tax law, such as:
| ● | banks, financial institutions or financial services entities; |
| ● | broker-dealers; |
| ● | taxpayers that are subject to the mark-to-market tax accounting rules; |
| ● | tax-exempt entities; |
| ● | governments or agencies or instrumentalities thereof; |
| ● | insurance companies; |
| ● | pension funds; |
| ● | mutual funds; |
| ● | regulated investment companies; |
| ● | real estate investment trusts; |
| ● | persons that acquired Holdco Ordinary Shares or Holdco Warrants pursuant to an exercise of employee share options, in connection with employee share incentive plans or otherwise as compensation; |
| ● | “specified foreign corporations” (including controlled foreign corporations), passive foreign investment companies or corporations that accumulate earnings to avoid U.S. federal income tax; |
| ● | tax-exempt organizations (including private foundations); |
| ● | persons that hold Holdco Ordinary Shares or Holdco Warrants as part of a “straddle,” “hedge,” “conversion,” “synthetic security,” “constructive ownership transaction,” “constructive sale,” “wash sale,” or other integrated or similar transaction for U.S. federal income tax purposes; |
| ● | persons that have a functional currency other than the U.S. dollar; |
| ● | U.S. expatriates or former long-term residents of the U.S.; |
| ● | persons owning or considered as owning (directly, indirectly, or through attribution) five percent (5%) (measured by vote or value) or more of the Holdco Ordinary Shares; |
| ● | persons who acquire Holdco Ordinary Shares as part of or in connection with the PIPE; |
| ● | accrual method taxpayers that file applicable financial statements as described in Section 451(b) of the Code; |
| ● | partnerships (or entities or arrangements classified as partnerships or other pass-through entities for U.S. federal income tax purposes, including S corporations) and any beneficial owners of such partnerships or other pass-through entities; and |
| ● | persons who are not U.S. Holders, all of whom may be subject to tax rules that differ materially from those summarized below. |
If a partnership (including an entity or arrangement treated as a partnership for U.S. federal income tax purposes) or other pass-through entity holds Holdco Ordinary Shares or Holdco Warrants, the tax treatment of a partner or other member in such partnership or other pass-through entity generally will depend upon the status of the partner or other member, the activities of the partnership or other pass-through entity and certain determinations made at the partner or member level. If you are a partner or member of a partnership or other pass-through entity holding Holdco Ordinary Shares or Holdco Warrants, you are urged to consult your tax advisor regarding the tax consequences to you of the ownership and disposition of Holdco Ordinary Shares and Holdco Warrants by the partnership or other pass-through entity.
176
This discussion is based on the Code, the regulations promulgated by the U.S. Treasury Department (“Treasury Regulations”), and judicial and administrative interpretations thereof, all as of the date hereof. All of the foregoing is subject to change, which change could apply retroactively and could affect the tax considerations described herein. Holdco has not sought, and does not intend to seek, any rulings from the Internal Revenue Service (the “IRS”) as to any U.S. federal income tax considerations described herein. Accordingly, there can be no assurance that the IRS will not take positions inconsistent with the considerations discussed below or that any such positions would not be sustained by a court.
THIS DISCUSSION IS ONLY A SUMMARY OF MATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONS ASSOCIATED WITH THE OWNERSHIP AND DISPOSITION OF HOLDCO ORDINARY SHARES AND HOLDCO WARRANTS. EACH HOLDER SHOULD CONSULT ITS OWN TAX ADVISOR WITH RESPECT TO THE PARTICULAR TAX CONSEQUENCES TO SUCH HOLDER OF THE OWNERSHIP AND DISPOSITION OF HOLDCO ORDINARY SHARES AND HOLDCO WARRANTS, INCLUDING THE APPLICABILITY AND EFFECTS OF U.S. FEDERAL, STATE AND LOCAL AND NON-U.S. TAX LAWS.
For purposes of this discussion, a “U.S. Holder” is a beneficial owner of Holdco Ordinary Shares or Holdco Warrants, as the case may be, that is:
| ● | an individual who is a U.S. citizen or resident of the United States; |
| ● | a corporation (including an entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof or the District of Columbia; |
| ● | an estate the income of which is includible in gross income for U.S. federal income tax purposes regardless of its source; or |
| ● | a trust (A) the administration of which is subject to the primary supervision of a U.S. court and which has one or more U.S. persons (within the meaning of the Code) who have the authority to control all substantial decisions of the trust or (B) that has in effect a valid election under applicable Treasury Regulations to be treated as a U.S. person. |
Tax Consequences of Ownership and Disposition of Holdco Ordinary Shares and Holdco Warrants
Dividends and Other Distributions on Holdco Ordinary Shares
Subject to the PFIC rules discussed below under the heading “— Passive Foreign Investment Company Rules,” distributions on Holdco Ordinary Shares generally will be taxable as a dividend for U.S. federal income tax purposes to the extent paid from Holdco’s current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Distributions in excess of Holdco’s current and accumulated earnings and profits will constitute a return of capital that will be applied against and reduce (but not below zero) the U.S. Holder’s adjusted tax basis in its Holdco Ordinary Shares. Any remaining excess will be treated as gain realized on the sale or other disposition of the Holdco Ordinary Shares and will be treated as described below under the heading “— Gain or Loss on Sale, Taxable Exchange or Other Taxable Disposition of Holdco Ordinary Shares and Holdco Warrants.” Holdco may not maintain calculations of earnings and profits under U.S. federal income tax principles for purposes of determining whether a distribution is a dividend for U.S. federal income tax purposes. Thus, it is possible that the full amount of any distributions will be reported as dividends for U.S. federal income tax purposes. The amount of any distribution will include any amounts withheld by Holdco (or another applicable withholding agent), which would include amounts expected to be payable in respect of Israeli income taxes, if any. Any amount treated as dividend income will be treated as foreign-source dividend income. Amounts treated as dividends that Holdco pays to a U.S. Holder that is a taxable corporation generally will be taxed at regular rates and will not qualify for the dividends received deduction generally allowed to domestic corporations in respect of dividends received from other domestic corporations. With respect to non-corporate U.S. Holders, under tax laws currently in effect and subject to certain exceptions (including, but not limited to, dividends treated as investment income for purposes of investment interest deduction limitations), dividends generally will be taxed at the lower applicable long-term capital gains rate only if the Holdco Ordinary Shares are readily tradable on an established securities market in the United States (which should include NASDAQ) or Holdco is eligible for benefits of the Convention between the Government of the United States of America and the Government of the State of Israel with respect to Taxes on Income (the “U.S.-Israel Tax Treaty”), and Holdco is not treated as a PFIC with respect to such U.S. Holder at the time the dividend was paid or in the preceding taxable year and provided certain holding period requirements are met. The amount of any dividend distribution paid in Israeli dollars will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars at that time. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt.
177
Subject to applicable limitations, Israeli income taxes withheld from dividends on the Holdco Ordinary Shares at a rate not exceeding the rate provided by the U.S.-Israel Tax Treaty generally will be eligible for credit against the U.S. treaty beneficiary’s U.S. federal income tax liability. The rules governing foreign tax credits are complex and U.S. Holders are urged to consult their tax advisers regarding the creditability of foreign taxes in their particular circumstances. In lieu of claiming a foreign tax credit, a U.S. Holder may deduct foreign taxes, including any Israeli income tax, in computing their taxable income, subject to generally applicable limitations under U.S. law. An election to deduct foreign taxes instead of claiming foreign tax credits applies to all foreign taxes paid or accrued in the taxable year.
Gain or Loss on Sale, Taxable Exchange or Other Taxable Disposition of Holdco Ordinary Shares and Holdco Warrants
Subject to the PFIC rules discussed below under the heading “— Passive Foreign Investment Company Rules,” upon any sale, exchange or other taxable disposition of any Holdco Ordinary Share or Holdco Warrant, a U.S. Holder generally will recognize gain or loss in an amount equal to the difference between (i) the sum of (x) the amount cash and (y) the fair market value of any other property, received in such sale, exchange or other taxable disposition and (ii) the U.S. Holder’s adjusted tax basis in such Holdco Ordinary Share or Holdco Warrant, as applicable (determined as described above or below), in each case as calculated in U.S. dollars. Any such gain or loss generally will be capital gain or loss and will be long-term capital gain or loss if the U.S. Holder’s holding period for such Holdco Ordinary Share or Holdco Warrant, as applicable, exceeds one year. Long-term capital gain realized by a non-corporate U.S. Holder generally will be taxable at a reduced rate. The deductibility of capital losses is subject to limitations.
This gain or loss generally will be treated as U.S. source gain or loss. Accordingly, in the event that any Israeli tax (including withholding tax) is imposed upon such sale, exchange or other taxable disposition, a U.S. Holder may not be able to utilize foreign tax credits unless such U.S. Holder has foreign source income or gain in the same category from other sources. In addition, there may be other limitations on utilizing foreign tax credits even if a U.S. Holder has foreign source income or gain in the same category from other sources. The rules governing foreign tax credits are complex and U.S. Holders are urged to consult their tax advisers regarding the creditability of foreign taxes in their particular circumstances.
If Holdco Ordinary Shares or Holdco Warrants are sold, exchanged, or otherwise disposed of in a taxable transaction for Israeli shekels, the amount realized generally will be the U.S. dollar value of the Israeli shekels received based on the spot rate in effect on the date of sale, exchange, or other taxable disposition. If a U.S. Holder is a cash method taxpayer and the Holdco Ordinary Shares or Holdco Warrants are traded on an established securities market, Israeli shekels received will be translated into U.S. dollars at the spot rate on the settlement date of the taxable disposition. An accrual method taxpayer may elect the same treatment with respect to the taxable disposition of Holdco Ordinary Shares or Holdco Warrants traded on an established securities market, provided that the election is applied consistently from year to year. Such election cannot be changed without the consent of the IRS. Israeli shekels received on the taxable disposition of a Holdco Ordinary Share or Holdco Warrant generally will have a tax basis equal to its U.S. dollar value as determined pursuant to the rules above. Any gain or loss recognized by a U.S. Holder on a sale, exchange, or other taxable disposition of the Israeli shekels will be ordinary income or loss and generally will be U.S.-source gain or loss.
Exercise, Lapse or Redemption of a Holdco Warrant
A U.S. Holder generally will not recognize gain or loss upon the acquisition of a Holdco Ordinary Share on the exercise of a Holdco Warrant for cash. A U.S. Holder’s tax basis in a Holdco Ordinary Share received upon exercise of the Holdco Warrant generally will equal the sum of the U.S. Holder’s tax basis in such Holdco Warrant and the exercise price. It is unclear whether a U.S. Holder’s holding period for the Holdco Ordinary Share received will commence on the date of exercise of the Holdco Warrant or the day following the date of exercise of the Holdco Warrant; in either case, the holding period will not include the period during which the U.S. Holder held the Holdco Warrant. If a Holdco Warrant is allowed to lapse unexercised, a U.S. Holder generally will recognize a capital loss equal to such holder’s tax basis in the Holdco Warrant.
The tax consequences of a cashless exercise of a warrant are not clear under current law. Subject to the PFIC rules discussed below, a cashless exercise may not be taxable, either because the exercise is not a realization event or because the exercise is treated as a recapitalization for U.S. federal income tax purposes. In either situation, a U.S. Holder’s tax basis in Holdco Ordinary Shares received generally should equal the U.S. Holder’s tax basis in the Holdco Warrants exercised therefor. If the cashless exercise was not a realization event, it is unclear whether a U.S. Holder’s holding period for the Holdco Ordinary Shares received would be treated as commencing on the date of exercise of the Holdco Warrants or the day following the date of exercise of the Holdco Warrants; in either case, the holding period will not include the period during which the U.S. Holder held the Holdco Warrants. If the cashless exercise were treated as a recapitalization, the holding period of the Holdco Ordinary Shares received would include the holding period of the Holdco Warrants.
178
It is also possible that a cashless exercise could be treated in part as a taxable exchange in which gain or loss would be recognized. In such event, a U.S. Holder could be deemed to have surrendered a number of Holdco Warrants equal to the number of Holdco Ordinary Shares having a value equal to the exercise price for the total number of Holdco Warrants to be exercised. In such case, subject to the PFIC rules discussed below, the U.S. Holder would recognize capital gain or loss with respect to the Holdco Warrants deemed surrendered in an amount equal to the difference between the fair market value of the Holdco Ordinary Shares that would have been received in a regular exercise of the Holdco Warrants deemed surrendered and the U.S. Holder’s tax basis in the Holdco Warrants deemed surrendered. In this case, a U.S. Holder’s aggregate tax basis in the Holdco Ordinary Shares received would equal the sum of the U.S. Holder’s tax basis in the Holdco Warrants deemed exercised and the aggregate exercise price of such Holdco Warrants. It is unclear whether a U.S. Holder’s holding period for the Holdco Ordinary Shares would commence on the date of exercise of the Holdco Warrants or the day following the date of exercise of the Holdco Warrants; in either case, the holding period will not include the period during which the U.S. Holder held the Holdco Warrants.
Due to the absence of authority on the U.S. federal income tax treatment of a cashless exercise, including when a U.S. Holder’s holding period would commence with respect to Holdco Ordinary Shares received, there can be no assurance regarding which, if any, of the alternative tax consequences and holding periods described above would be adopted by the IRS or a court of law. Accordingly, U.S. Holders should consult their tax advisors regarding the tax consequences of a cashless exercise.
Subject to the PFIC rules described below, if Holdco redeems Holdco Warrants for cash pursuant to the redemption provisions described in Exhibit 2.1 to this Annual Report or if Holdco purchases Holdco Warrants in an open market transaction, such redemption or purchase generally will be treated as a taxable disposition to the U.S. Holder, taxed as described above under “—Gain or Loss on Sale, Taxable Exchange or Other Taxable Disposition of Holdco Ordinary Shares and Holdco Warrants.”
Possible Constructive Distributions
The terms of each Holdco Warrant provide for an adjustment to the number of Holdco Ordinary Shares for which the Holdco Warrant may be exercised or to the exercise price of the Holdco Warrant in certain events, as discussed in Exhibit 2.1 to this Annual Report. An adjustment which has the effect of preventing dilution generally is not taxable. U.S. Holders of Holdco Warrants would, however, be treated as receiving a constructive distribution from Holdco if, for example, the adjustment increases such U.S. Holders’ proportionate interest in Holdco’s assets or earnings and profits (e.g., through an increase in the number of Holdco Ordinary Shares that would be obtained upon exercise or through a decrease in the exercise price of the Holdco Warrants), which adjustment may be made as a result of a distribution of cash or other property to the holders of Holdco Ordinary Shares. Such constructive distribution to a U.S. Holder of Holdco Warrants would be treated as if such U.S. Holder had received a cash distribution from Holdco generally equal to the fair market value of such increased interest (taxed as described above under “—Dividends and Other Distributions on Holdco Ordinary Shares”).
Passive Foreign Investment Company Rules
The treatment of U.S. Holders of Holdco Ordinary Shares and Holdco Warrants could be materially different from that described above if Holdco is treated as a PFIC for U.S. federal income tax purposes.
A foreign (i.e., non-U.S.) corporation will be classified as a PFIC for U.S. federal income tax purposes if either (i) at least 75% of its gross income in a taxable year, including its pro rata share of the gross income of any corporation in which it is considered to own at least 25% of the shares by value, is passive income or (ii) at least 50% of its assets in a taxable year (ordinarily determined based on fair market value and averaged quarterly over the year), including its pro rata share of the assets of any corporation in which it is considered to own at least 25% of the shares by value, are held for the production of, or produce, passive income. Passive income generally includes dividends, interest, rents and royalties (other than rents or royalties derived from the active conduct of a trade or business) and gains from the disposition of passive assets. Cash may be treated as a passive asset for this purpose. Pursuant to the start-up exception, a corporation will not be a PFIC for the first taxable year the corporation has gross income (the “Start-Up Year”), if (1) no predecessor of the corporation was a PFIC; (2) the corporation establishes to the satisfaction of the IRS that it will not be a PFIC for either of the first two taxable years following the Start-Up Year; and (3) the corporation is not in fact a PFIC for either of those years (the “Start-Up Exception”).
Because LAMF was a blank-check company with no current active business, based upon the composition of LAMF’s income and assets for its first taxable year (ending December 31, 2021) and its second taxable year (ending December 31, 2022), LAMF stated that it believed it did not qualify for the Start-Up Exception and believes it was a PFIC for such taxable years. In addition, because LAMF was a blank-check company with no current active business, based upon the expected composition of LAMF’s income and assets for its taxable year ending December 31, 2023, LAMF expects to be a PFIC for such taxable year.
179
As a result of the Business Combination, Holdco should be treated as LAMF’s successor for U.S. federal income tax purposes and LAMF’s current taxable year will not close and will continue under Holdco. Thus, for purposes of the PFIC rules, Holdco Ordinary Shares generally will be treated as the LAMF Class A Ordinary Shares exchanged in the SPAC Merger, and Holdco Warrants generally will be treated as the Public Warrants exchanged in the SPAC Merger. Following the Business Combination, the annual PFIC income and asset tests in respect of Holdco will be applied based on the assets and activities of the combined business. To determine whether the PFIC asset test has been met, a calendar-year corporation generally divides the average of the values of passive assets at the end of each quarter by the average value of all assets at the end of each quarter. Based on the projected composition of Holdco’s income and assets, there is a significant risk that Holdco will be classified as a PFIC for its taxable year that includes the date of the Business Combination and in the foreseeable future.
However, because PFIC status is based on income, assets and activities for the entire taxable year, it is not possible to determine the PFIC status of Holdco for any taxable year until after the close of the taxable year.
It is not entirely clear how various aspects of the PFIC rules apply to the warrants. Section 1298(a)(4) of the Code provides that, to the extent provided in Treasury Regulations, any person who has an option to acquire stock in a PFIC shall be considered to own such stock in the PFIC for purposes of the PFIC rules. No final Treasury regulations are currently in effect under Section 1298(a)(4) of the Code. However, proposed Treasury Regulations under Section 1298(a)(4) of the Code have been promulgated with a retroactive effective date (the “Proposed PFIC Option Regulations”). Each U.S. Holder is urged to consult its tax advisors regarding the possible application of the Proposed PFIC Option Regulations to an investment in the Holdco Warrants. Solely for discussion purposes, the following discussion assumes that the Proposed PFIC Option Regulations will apply to the Holdco Warrants.
Although the PFIC status of Holdco is determined annually, an initial determination that Holdco (or, before the Business Combination, LAMF) is a PFIC generally will apply for subsequent years to a U.S. Holder who held shares or warrants in such company while such company was a PFIC, whether or not such company meets the test for PFIC status in those subsequent years. If Holdco is determined to be a PFIC (for any taxable year) with respect to a U.S. Holder, and such U.S. Holder did not timely make any of the PFIC Elections with respect to such shares or warrants, such U.S. Holder generally will be subject to special rules with respect to (i) any gain recognized by the U.S. Holder on the sale or other disposition of its Holdco Ordinary Shares and Holdco Warrants (which may include gain realized by reason of transfers of such shares or warrants that would otherwise qualify as nonrecognition transactions for U.S. federal income tax purposes) and (ii) any “excess distribution” made to the U.S. Holder (generally, any distributions to such U.S. Holder during a taxable year of the U.S. Holder that are greater than 125% of the average annual distributions received by such U.S. Holder in respect of the Holdco Ordinary Shares during the three preceding taxable years of such U.S. Holder or, if shorter, the portion of such U.S. Holder’s holding period for such shares that preceded the taxable year of the distribution) (together, the “excess distribution rules”).
Under these excess distribution rules:
| ● | the U.S. Holder’s gain or excess distribution will be allocated ratably over the U.S. Holder’s holding period for the Holdco Ordinary Shares and Holdco Warrants; |
| ● | the amount allocated to the U.S. Holder’s taxable year in which the U.S. Holder recognized the gain or received the excess distribution, or to the period in the U.S. Holder’s holding period before the first day of Holdco’s first taxable year in which Holdco is a PFIC, will be taxed as ordinary income; |
| ● | the amount allocated to other taxable years (or portions thereof) of the U.S. Holder and included in its holding period will be taxed at the highest tax rate in effect for that year and applicable to the U.S. Holder without regard to the U.S. Holder’s other items of income and loss for such year; and |
| ● | an additional amount equal to the interest charge generally applicable to underpayments of tax will be imposed on the U.S. Holder with respect to the tax attributable to each such other taxable year of the U.S. Holder. |
In general, if Holdco is determined to be a PFIC, a U.S. Holder may be able to avoid the excess distribution rules described above with respect to Holdco Ordinary Shares (but, under current law, not the Holdco Warrants) by making (or having made) a timely and valid QEF election (if eligible to do so) to include in income its pro rata share of Holdco’s net capital gains (as long-term capital gain) and other earnings and profits (as ordinary income), on a current basis, in each case whether or not distributed, in the taxable year of the U.S. Holder in which or with which Holdco’s taxable year ends. A U.S. Holder generally may make a separate election to defer the payment of taxes on undistributed income inclusions under the QEF rules, but if deferred, any such taxes will be subject to an interest charge.
180
If a U.S. Holder makes a QEF election with respect to its Holdco Ordinary Shares in a year after Holdco’s first taxable year as a PFIC in which the U.S. Holder held (or was deemed to hold) Holdco Ordinary Shares, then notwithstanding such QEF election, the excess distribution rules discussed above, adjusted to take into account the current income inclusions resulting from the QEF election, will continue to apply with respect to such U.S. Holder’s Holdco Ordinary Shares, unless the U.S. Holder makes a purging election under the PFIC rules. Under one type of purging election, the U.S. Holder will be deemed to have sold such Holdco Ordinary Shares at their fair market value and any gain recognized on such deemed sale will be treated as an excess distribution, as described above. As a result of such purging election, the U.S. Holder will have additional basis (to the extent of any gain recognized on the deemed sale) and, solely for purposes of the PFIC rules, a new holding period in the Holdco Ordinary Shares.
Under current law, a U.S. Holder may not make a QEF election with respect to its Holdco Warrants. As a result, if a U.S. Holder sells or otherwise disposes of such warrants (other than upon exercise of such warrants) and Holdco were a PFIC at any time during the U.S. Holder’s holding period of such warrants, any gain recognized generally will be treated as an excess distribution, taxed as described above. If a U.S. Holder that exercises such warrants properly makes and maintains a QEF election with respect to the newly acquired Holdco Ordinary Shares (or has previously made a QEF election with respect to Holdco Ordinary Shares), the QEF election will apply to the newly acquired Holdco Ordinary Shares. Notwithstanding such QEF election, the excess distribution rules discussed above, adjusted to take into account the current income inclusions resulting from the QEF election, will continue to apply with respect to such newly acquired Holdco Ordinary Shares (which, while not entirely clear, generally will be deemed to have a holding period for purposes of the PFIC rules that includes the period the U.S. Holder held the Holdco Warrants), unless the U.S. Holder makes a purging election under the PFIC rules. U.S. Holders are urged to consult their tax advisors as to the application of the rules governing purging elections to their particular circumstances.
The QEF election is made on a shareholder-by-shareholder basis and, once made, can be revoked only with the consent of the IRS. A U.S. Holder generally makes a QEF election by attaching a completed IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund), including the information provided in a PFIC Annual Information Statement, to a timely filed United States federal income tax return for the tax year to which the election relates. Retroactive QEF elections generally may be made only by filing a protective statement with such return and if certain other conditions are met or with the consent of the IRS. U.S. Holders should consult their tax advisors regarding the availability and tax consequences of a retroactive QEF election under their particular circumstances.
In order to comply with the requirements of a QEF election, a U.S. Holder must receive a PFIC Annual Information Statement from Holdco. In general, if Holdco determines that it was a PFIC in any taxable year, Holdco will use commercially reasonable efforts to provide the statements and information (including a PFIC Annual Information Statement) necessary to enable U.S. Holders to make and comply with the requirements of QEF elections or to file a protective statement. However, there is no assurance that Holdco will timely provide such required information. There is also no assurance that Holdco will have timely knowledge of its status as a PFIC in the future or of such information in order for U.S. Holders to make or maintain a QEF election.
If a U.S. Holder has made a QEF election with respect to the Holdco Ordinary Shares, and the excess distribution rules discussed above do not apply to such shares (because of a timely QEF election for Holdco’s first taxable year as a PFIC in which the U.S. Holder holds (or is deemed to hold) such shares or a purge of the PFIC taint pursuant to a purging election, as described above), any gain recognized on the sale of Holdco Ordinary Shares generally will be taxable as capital gain and no additional interest charge will be imposed under the PFIC rules. As discussed above, if Holdco is a PFIC for any taxable year, a U.S. Holder of Holdco Ordinary Shares that has made a QEF election will be currently taxed on its pro rata share of Holdco’s earnings and profits, whether or not distributed for such year. A subsequent distribution of such earnings and profits that were previously included in income generally should not be taxable when distributed to such U.S. Holder. The tax basis of a U.S. Holder’s shares in a QEF will be increased by amounts that are included in income, and decreased by amounts distributed but not taxed as dividends, under the above rules. In addition, if Holdco is not a PFIC for any taxable year, such U.S. Holder will not be subject to the QEF inclusion regime with respect to Holdco Ordinary Shares for such a taxable year.
Alternatively, if a U.S. Holder, at the close of its taxable year, owns shares in a PFIC that are treated as marketable stock, the U.S. Holder may make a mark-to-market election with respect to such shares for such taxable year. If the U.S. Holder makes a valid mark-to-market election for the first taxable year of the U.S. Holder in which the U.S. Holder holds (or is deemed to hold) Holdco Ordinary Shares and for which Holdco is determined to be a PFIC, such U.S. Holder generally will not be subject to the excess distribution rules described above with respect to its Holdco Ordinary Shares. Instead, in general, the U.S. Holder will include as ordinary income in each taxable year the excess, if any, of the fair market value of its Holdco Ordinary Shares at the end of its taxable year over its adjusted basis in its Holdco Ordinary Shares. These amounts of ordinary income would not be eligible for the favorable tax rates applicable to qualified dividend income or long-term capital gains. The U.S. Holder also will recognize an ordinary loss in respect of the excess, if any, of its adjusted basis in its Holdco Ordinary Shares over the fair market value of its Holdco Ordinary Shares at the end of its taxable year (but only to the extent of the net amount of previously included income as a result of the mark-to-market election). The U.S. Holder’s basis in its Holdco Ordinary Shares will be adjusted to reflect any such income or loss amounts, and any further gain recognized on a sale or other taxable disposition of its Holdco Ordinary Shares will be treated as ordinary income. Under current law, a mark-to-market election may not be made with respect to warrants.
181
The mark-to-market election is available only for stock that is regularly traded on a national securities exchange that is registered with the Securities and Exchange Commission, including Nasdaq (on which the Holdco Ordinary Shares are intended to be listed), or on a foreign exchange or market that the IRS determines has rules sufficient to ensure that the market price represents a legitimate and sound fair market value. If made, a mark-to-market election would be effective for the taxable year for which the election was made and for all subsequent taxable years unless the Holdco Ordinary Shares ceased to qualify as “marketable stock” for purposes of the PFIC rules or the IRS consented to the revocation of the election. U.S. Holders are urged to consult their own tax advisors regarding the availability and tax consequences of a mark-to-market election with respect to Holdco Ordinary Shares under their particular circumstances.
If Holdco is a PFIC and, at any time, has a non-U.S. subsidiary that is classified as a PFIC, U.S. Holders generally would be deemed to own a portion of the shares of such lower-tier PFIC, and generally could incur liability for the deferred tax and interest charge described above if Holdco receives a distribution from, or dispose of all or part of Holdco’s interest in, the lower-tier PFIC or the U.S. Holders otherwise were deemed to have disposed of an interest in the lower-tier PFIC. In general, if Holdco determines that it was a PFIC in any taxable year, Holdco will use commercially reasonable efforts to cause any lower-tier PFIC to provide to a U.S. Holder the information that may be required to make or maintain a QEF election with respect to the lower-tier PFIC. There can be no assurance that Holdco will have timely knowledge of the status of any such lower-tier PFIC. In addition, Holdco may not hold a controlling interest in any such lower-tier PFIC and thus there can be no assurance that Holdco will be able to cause the lower-tier PFIC to provide such information. A mark-to-market election generally would not be available with respect to such lower-tier PFIC. U.S. Holders are urged to consult their tax advisors regarding the tax issues raised by lower-tier PFICs.
A U.S. Holder that owns (or is deemed to own) shares in a PFIC during any taxable year of the U.S. Holder may have to file an IRS Form 8621 (whether or not a QEF or mark-to-market election is made) and such other information as may be required by the U.S. Treasury Department. Failure to do so, if required, will extend the statute of limitations until such required information is furnished to the IRS.
The rules dealing with PFICs and PFIC Elections are very complex and are affected by various factors in addition to those described above. Accordingly, U.S. Holders of Holdco Ordinary Shares and Holdco Warrants should consult their own tax advisors concerning the application of the PFIC rules to Holdco Ordinary Shares and Holdco Warrants under their particular circumstances.
Additional Reporting Requirements
Certain U.S. Holders may be required to file an IRS Form 926 (Return by a U.S. Transferor of Property to a Foreign Corporation) to report a transfer of property (including cash) to Holdco. Substantial penalties may be imposed on a U.S. Holder that fails to comply with this reporting requirement, and the period of limitations on assessment and collection of U.S. federal income taxes will be extended in the event of a failure to comply. Furthermore, certain U.S. Holders who are individuals and certain entities will be required to report information with respect to such U.S. Holder’s investment in “specified foreign financial assets” on IRS Form 8938 (Statement of Specified Foreign Financial Assets), subject to certain exceptions. Specified foreign financial assets generally include any financial account maintained with a non-U.S. financial institution and should also include Holdco Ordinary Shares and Holdco Warrants if they are not held in an account maintained with a U.S. financial institution. Persons who are required to report specified foreign financial assets and fail to do so may be subject to substantial penalties, and the period of limitations on assessment and collection of U.S. federal income taxes may be extended in the event of a failure to comply. U.S. Holders are urged to consult their tax advisors regarding the foreign financial asset and other reporting obligations and their application to an investment in Holdco Ordinary Shares and Holdco Warrants.
Treasury Regulations meant to require the reporting of certain tax shelter transactions could be interpreted to cover transactions generally not regarded as tax shelters, including certain foreign currency transactions. Under the applicable Treasury Regulations, certain transactions are required to be reported to the IRS including, in certain circumstances, a sale, exchange, retirement or other taxable disposition of foreign currency, to the extent that such sale, exchange, retirement or other taxable disposition results in a tax loss in excess of a threshold amount. You should consult your tax advisor to determine the tax return obligations, if any, with respect to Holdco Ordinary Shares, Holdco Warrants, and the receipt of Israeli shekels in respect thereof, including any requirement to file IRS Form 8886 (Reportable Transaction Disclosure Statement).
182
Information Reporting and Backup Withholding
Dividend payments with respect to Holdco Ordinary Shares and proceeds from the sale, exchange or redemption of Holdco Ordinary Shares or Holdco Warrants may be subject to information reporting to the IRS and possible United States backup withholding. Backup withholding will not apply, however, to a U.S. Holder who furnishes a correct taxpayer identification number and makes other required certifications, or who is otherwise exempt from backup withholding and establishes such exempt status.
Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against a U.S. Holder’s United States federal income tax liability, and a U.S. Holder generally may obtain a refund of any excess amounts withheld under the backup withholding rules by timely filing the appropriate claim for refund with the IRS and furnishing any required information.
Material Israeli Tax Considerations
General Corporate Tax Structure in Israel
Generally, Israeli companies are subject to corporate tax on their taxable income. Since 2018, the corporate tax rate has been 23%. However, the effective tax rate payable by a company that derives income from an “Approved Enterprise”, a “Beneficiary Enterprise” or a “Preferred Enterprise”, a “Special Preferred Enterprise”, a “Preferred Technology Enterprise” or “Special Preferred Technology Enterprise”, as those terms are defined under the Law for the Encouragement of Capital Investments, 5719-1959, may be considerably lower. Capital gains derived by an Israeli company are generally subject to the prevailing regular corporate tax rate.
Holdco does not enjoy the tax benefits under the Law for the Encouragement of Capital Investments, 5719-1959, and therefore is subject to general corporate tax on its taxable income at the rate of 23%.
Tax benefits and grants for research and development
Israeli tax law allows, under certain conditions, a tax deduction for expenditures, including capital expenditures, for the year in which they are incurred. Expenditures are deemed related to scientific research and development projects, if:
| ● | The expenditures are approved by the relevant Israeli government ministry, determined by the field of research; |
| ● | The research and development must be for the promotion of the company; and |
| ● | The research and development is carried out by or on behalf of the company seeking such tax deduction. |
The amount of such deductible expenses is reduced by the sum of any funds received through government grants for the finance of such scientific research and development projects. No deduction under these research and development deduction rules is allowed if such deduction is related to an expense invested in an asset depreciable under the general depreciation rules of the Israeli Income Tax Ordinance (New Version) 1961, referred to as the Ordinance. Expenditures that are not qualified under the conditions above are deductible in equal amounts over a three-year period.
183
Israeli Taxation Considerations for Holdco Shareholders
Israeli law generally imposes a capital gains tax on the sale of any capital assets by residents of Israel, as defined for Israeli tax purposes, and on the sale of assets located in Israel, including shares of Israeli companies, by both residents and non-residents of Israel unless a specific exemption is available or unless a tax treaty between Israel and the seller’s country of residence provides otherwise. The Ordinance distinguishes between “Real Capital Gain” and “Inflationary Surplus”. The Real Capital Gain is the excess of the total capital gain over the Inflationary Surplus. The Inflationary Surplus is a portion of the total capital gain which is equivalent to the increase of the relevant asset’s purchase price which is attributable to the increase in the Israeli consumer price index or, in certain circumstances, a foreign currency exchange rate, between the date of purchase and the date of sale. Inflationary Surplus is not subject to tax in Israel.
Israeli Resident Individuals
Capital Gain
As of January 1, 2006, the tax rate applicable to Real Capital Gain derived by Israeli individuals from the sale of shares which had been purchased on or after January 1, 2003, whether or not listed on a stock exchange, is 20%, unless such shareholder claims a deduction for interest and linkage differences expenses in connection with the purchase and holding of such shares, in which case the gain will generally be taxed at a rate of 25%. Additionally, if such shareholder is considered a “Significant Shareholder” (i.e., a person who holds, directly or indirectly, alone or together with another person who collaborates with such person on a permanent basis, 10% or more of any of the company’s “means of control” (including, among other things, the right to receive profits of the company, voting rights, the right to receive the company’s liquidation proceeds and the right to appoint a director)) at the time of sale or at any time during the preceding 12-month period, such gain will be taxed at the rate of 25%.
Notwithstanding the foregoing, pursuant to the Law for Change in the Tax Burden (Legislative Amendments) (Taxes), 2011, the capital gain tax rate applicable to individuals was raised from 20% to 25% from 2012 and onwards (or from 25% to 30% if the selling individual shareholder is a Significant Shareholder at any time during the 12-month period preceding the sale and/or claims a deduction for interest and linkage differences expenses in connection with the purchase and holding of such shares). With respect to assets (not shares that are listed on a stock exchange) purchased on or after January 1, 2003, the portion of the gain generated from the date of acquisition until December 31, 2011 will be subject to the previous capital gains tax rates (20% or 25%) and the portion of the gain generated from January 1, 2012 until the date of sale will be subject to the new tax rates (25% or 30%).
Individual shareholders dealing in securities in Israel are taxed at their marginal tax rates applicable to business income (up to 47% in 2023, excluding, excess tax, if any, as described below) unless the benefiting provisions of an applicable treaty applies.
Dividend Income
Israeli residents who are individuals are generally subject to Israeli income tax for dividends paid on our ordinary shares (other than bonus shares or share dividends) at 25%, or 30% if the recipient of such dividend is a Significant Shareholder, at the time of distribution or at any time during the preceding 12-month period.
Israeli Resident Corporations
Capital Gain
Under current Israeli tax legislation, the tax rate applicable to Real Capital Gain derived by Israeli resident corporations from the sale of shares of an Israeli company is the general corporate tax rate. The general corporate tax rate has been 23% since 2018.
Dividend Income
Generally, Israeli resident corporations are exempt from Israeli corporate tax on the receipt of dividends paid on shares of Israeli resident corporations.
184
Non-Israeli Residents
Capital Gain
Israeli capital gains tax is imposed on the disposal of capital assets by a non-Israeli resident if such assets are either (i) located in Israel; (ii) shares or rights to shares in an Israeli resident company, or (iii) represent, directly or indirectly, rights to assets located in Israel, unless a tax treaty between Israel and the seller’s country of residence provides otherwise. As mentioned above, Real Capital Gain is generally subject to tax at the general corporate tax rate (23% since 2018), if generated by a company, or at the rate of 25% (for any asset other than shares that are listed on a stock exchange, 20% with respect to the portion of the gain generated up to December 31, 2011) or 30% (for any asset other than shares that are listed on a stock exchange, 25% with respect to the portion of the gain generated up to December 31, 2011), if generated by an individual who is Significant Shareholder at the time of sale or at any time during the preceding 12-month period (or claims a deduction for interest and linkage differences expenses in connection with the purchase and holding of such shares) from the sale of assets purchased on or after January 1, 2003.
Individual and corporate shareholders dealing in securities in Israel are taxed at the tax rates applicable to business income (a corporate tax rate for a corporation and a marginal tax rate of up to 47% for an individual in 2023) unless contrary provisions in a relevant tax treaty applies.
Notwithstanding the foregoing, shareholders who are non-Israeli residents (individuals and corporations) should generally exempt from Israeli capital gains tax on any gains derived from the sale, exchange or disposition of shares publicly traded on a recognized stock exchange outside of Israel, provided, among other things, that such gains are not generated through a permanent establishment that the non-Israeli resident maintains in Israel, and the shares were purchased after January 1, 2009 or after the Company was listed on a recognized stock exchange. However, non-Israeli corporations will not be entitled to the foregoing exemptions if Israeli residents (a) have a controlling interest of more than 25% in such non-Israeli corporation, or (b) are the beneficiaries of or are entitled to 25% or more of the revenues or profits of such non-Israeli corporation, whether directly or indirectly. Such exemption is not applicable to a person whose gains from selling or otherwise disposing of the shares are deemed to be business income.
In addition, a sale of shares may be exempt from Israeli capital gains tax under the provisions of an applicable tax treaty. For example, under the U.S.-Israel Tax Treaty, or U.S.-Israel Treaty, the sale, exchange or disposition of shares of an Israeli company by a shareholder who is a U.S. resident (for purposes of the U.S.-Israel Treaty) holding the shares as a capital asset and is entitled to claim the benefits afforded to such a resident by the U.S.-Israel Tax Treaty is generally exempt from Israeli capital gains tax unless either (i) the shareholder holds, directly or indirectly, shares representing 10% or more of the voting rights during any part of the 12-month period preceding such sale, exchange or disposition, subject to certain conditions; (ii) the shareholder, if an individual, has been present in Israel for a period or periods of 183 days or more in the aggregate during the applicable taxable year; (iii) the capital gains arising from such sale, exchange or disposition are attributable to a permanent establishment of the shareholder which is maintained in Israel, under certain terms; (iv) the capital gain arising from such sale, exchange or disposition is attributed to real estate located in Israel, or (v) the capital gains arising from such sale, exchange or disposition is attributed to royalties. In any such case, the sale, exchange or disposition of such shares would be subject to Israeli tax, to the extent applicable; however, under the U.S.-Israel Treaty, a U.S. resident would be permitted to claim a credit for the Israeli tax against the U.S. federal income tax imposed with respect to the sale, exchange or disposition, subject to the limitations in U.S. laws applicable to foreign tax credits. The U.S.-Israel Treaty does not provide such credit against any U.S. state or local taxes.
In some instances where our shareholders may be liable for Israeli tax on the sale of their shares, the payment of the consideration may be subject to the withholding of Israeli tax at source. Shareholders may be required to demonstrate that they are exempt from tax on their capital gains in order to avoid withholding at source at the time of sale. Specifically, in transactions involving a sale of all of the shares of an Israeli resident company, in the form of a merger or otherwise, the Israel Tax Authority may require from shareholders who are not liable for Israeli tax to sign declarations in forms specified by this authority or obtain a specific exemption from the Israel Tax Authority to confirm their status as non-Israeli resident, and, in the absence of such declarations or exemptions, may require the purchaser of the shares to withhold taxes at source.
185
Dividend Income
Non-Israeli residents (whether individuals or corporations) are generally subject to Israeli income tax on the receipt of dividends paid on shares at the rate of 25% or 30% (if the dividend recipient is a Significant Shareholder at the time of distribution or at any time during the preceding 12-month period). Such dividends are generally subject to Israeli withholding tax at a rate of 25% so long as the shares are registered with a nominee company (whether the recipient is a Significant Shareholder or not), or such lower rate as may be provided in an applicable tax treaty. For example, under the U.S.-Israel Treaty, the maximum rate of tax withheld at source in Israel on dividends paid to a holder of our ordinary shares who is a U.S. resident (for purposes of the U.S.-Israel Treaty) is 25%. However, generally, the maximum rate of withholding tax on dividends, that are paid to a U.S. corporation holding at least 10% or more of our outstanding voting capital from the start of the tax year preceding the distribution of the dividend through (and including) the distribution of the dividend, is 12.5%, provided that no more than 25% of our gross income for such preceding year consists of certain types of dividends and interest. The aforementioned rates will not apply if the dividend income was generated through a permanent establishment of the U.S. resident that is maintained in Israel. U.S. residents who are subject to Israeli withholding tax on a dividend may be entitled to a credit or deduction for United States federal income tax purposes in the amount of the taxes withheld, subject to detailed rules contained in the Code.
A non-Israeli resident who receives dividends from which tax was withheld is generally exempt from the obligation to file tax returns in Israel with respect to such income, provided that (i) such income was not generated from business conducted in Israel by the taxpayer, (ii) the taxpayer has no other taxable sources of income in Israel with respect to which a tax return is required to be filed, and (iii) the taxpayer is not obligated to pay excess tax (as further explained below).
Excess Tax
Subject to the provisions of an applicable tax treaty, individuals who are subject to tax in Israel (whether any such individual is an Israeli resident or non-Israeli resident) are also subject to an additional tax at a rate of 3% on annual income (including, but not limited to, income derived from dividends, interest and capital gain) exceeding NIS 721,560 for 2024, which amount is linked to the annual change in the Israeli consumer price index.
| F. | Dividends and Paying Agents |
Not applicable.
| G. | Statement by Experts |
Not applicable.
| H. | Documents on Display |
We are subject to certain of the informational filing requirements of the Exchange Act. Since we are a “foreign private issuer,” we are exempt from the rules and regulations under the Exchange Act prescribing the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions contained in Section 16 of the Exchange Act, with respect to their purchase and sale of our equity securities. In addition, we are not required to file reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. However, we are required to file with the SEC an Annual Report on Form 20-F containing financial statements audited by an independent accounting firm. We will also furnish to the SEC, on Form 6-K, unaudited financial information with respect to our interim results. Information filed with or furnished to the SEC by us will be available on our website. The SEC also maintains a website at http://www.sec.gov that contains reports and other information that we file with or furnish electronically with the SEC.
| I. | Subsidiary Information |
Not applicable.
| J. | Annual Report to Security Holders |
Not applicable.
186
| ITEM 11. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Interest Rate Risk
We are exposed to market risks in the ordinary course of our business. These risks primarily relate to interest rates. As of December 31, 2023, we had cash and cash equivalents of $0.55 million, all of which was held in checking accounts. The $6.83 million of principal outstanding on our Nuvo Convertible Loans bears interest at a fixed rate of 2% per month and the $2.1 million of principal outstanding on our Nuvo Bridge Loans bears interest at a fixed rate of 15% per year as described above. We therefore do not believe we are exposed to, nor do we anticipate being in the near future exposed to, material risks due to changes in interest rates.
Inflation-Related Risks
We do not believe that the rate of inflation in Israel has had a material impact on our business to date. However, our costs in Israel will increase if the inflation rate in Israel exceeds the devaluation of the NIS against the U.S. dollar or if the timing of such devaluation lags behind inflation in Israel. In any such event, the dollar cost of our operations in Israel would increase and our dollar-denominated results of operations would be adversely affected. We cannot predict any future trends in the rate of inflation in Israel or the rate of devaluation (if any) of the NIS against the dollar. To the extent inflation increases our costs and expenses, we may have to consider price increases to offset those cost pressures.
Foreign Currency Exchange Risk
Our foreign currency exposures give rise to market risk associated with exchange rate movements of the NIS mainly against the U.S. dollar, and vice versa, because most of our expenses are denominated in NIS and the U.S. dollar. Our NIS and U.S. dollar expenses consist principally of payments made to employees, subcontractors and consultants for preclinical studies, clinical trials and other research and development activities. We anticipate that a sizable portion of our expenses will continue to be denominated in the NIS and U.S. dollar. Our financial position, results of operations and cash flows are, therefore, subject to fluctuations due to changes in foreign currency exchange rates and may be adversely affected in the future due to changes in foreign exchange rates.
| ITEM 12. | DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES |
Not applicable.
187
PART II
| ITEM 13. | DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES |
None.
| ITEM 14. | MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS |
None.
| ITEM 15. | CONTROLS AND PROCEDURES |
Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of December 31, 2023. Based on such evaluation, those officers concluded that, as of December 31, 2023, our disclosure controls and procedures were effective in recording, processing, summarizing and reporting, on a timely basis, information required to be included in periodic filings under the Exchange Act and that such information is accumulated and communicated to management, including our principal executive and financial officers, as appropriate to allow timely decisions regarding required disclosure.
This Annual Report does not include a report of management’s assessment regarding internal control over financial reporting or an attestation report of the Company’s registered public accounting firm due to a transition period established by rules of the Securities and Exchange Commission for newly public companies.
During the year ended December 31, 2023, there were no changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| ITEM 16. | [RESERVED] |
| ITEM 16A. | AUDIT COMMITTEE FINANCIAL EXPERT |
Our Board of Director has determined that Christina Spade, a member of our Audit Committee, is an audit committee financial expert, as defined under the rules under the Exchange Act, and is independent in accordance with applicable Exchange Act rules and Nasdaq rules.
| ITEM 16B. | CODE OF ETHICS |
We have adopted a written code of business conduct (the “Code of Business Conduct”) that applies to all directors, executive officers and employees. Our Code of Business Conduct is available on our website at https://www.nuvocares.com. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report on Form 20-F and is not incorporated by reference herein. If we make any amendment to the Code of Business Conduct or grant any waivers, including any implicit waiver, from a provision of the code, we will disclose the nature of such amendment or waiver on our website to the extent required by the rules and regulations of the SEC including the instructions to Item 16B of Form 20-F. We have not granted any waivers under our Code of Business Conduct.
188
| ITEM 16C. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The following table provides information regarding fees paid by us to Kesselman & Kesselman, Certified Public Accountant, a member of PricewaterhouseCoopers International Limited, our principal independent registered public accounting firm, for all services performed for Nuvo, including audit services, for the years ended December 31, 2023 and 2022, as well as the fees for services performed for Holdco for the preparation of the audited consolidated financial statements of Holdco as of December 31, 2023 and July 20, 2023:
| Year Ended December 31, |
||||||||
| 2023 | 2022 | |||||||
| Audit fees (1) | $ | 418,142 | $ | 329,600 | ||||
| Audit related fees | - | - | ||||||
| Tax Fees (2) | 38,808 | - | ||||||
| All other fees | - | - | ||||||
| Total | $ | 456,950 | $ | 329,600 | ||||
| (1) | Includes professional services rendered in connection with the audit of our annual financial statements, the review of our interim financial statements, statutory audits and includes audit services related to the de-SPAC transaction, consents, and assistance with review of documents filed with the SEC. |
| (2) | Includes fees related to professional services in connection with tax compliance services and other tax related services. |
Pre-Approval of Auditors’ Compensation
All of the audit services, audit-related services and tax services described in the table with respect to Nuvo above were approved in advance by Nuvo’s Audit Committee prior to forming the Holdco Nuvo Group D.G Ltd. public company Audit Committee in accordance with paragraph (c)(7)(i)(A) of Rule 2-01 of Regulation S-X and thereafter approved by our Board of Directors in accordance with Israeli law.
| ITEM 16D. | EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES |
Not applicable.
| ITEM 16E. | PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS |
Repurchases, if any, may be effected from time to time through open market purchases, including pursuant to a pre-set trading plan meeting the requirements of Rule 10b5-1(c) of the Exchange Act, privately negotiated transactions, pursuant to accelerated share repurchase agreements entered into with one or more counterparties, or otherwise.
During the year ended December 31, 2023, neither we nor any affiliated purchaser (as defined in the Exchange Act) purchased any of our ordinary shares.
| ITEM 16F. | CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT |
Not applicable.
189
| ITEM 16G. | CORPORATE GOVERNANCE |
As an Israeli company, we are subject to various corporate governance requirements under the Companies Law. However, pursuant to regulations promulgated under the Companies Law, companies with shares traded on certain U.S. stock exchanges, including Nasdaq, may, subject to certain conditions, “opt out” from the Companies Law requirements to appoint external directors and related Companies Law rules concerning the composition of the audit committee and compensation committee of the board of directors (other than the gender diversification rule under the Companies Law, which requires the appointment of a director from the other gender if at the time a director is appointed all members of the board of directors are of the same gender). In accordance with these regulations, we elected to “opt out” from those requirements of the Companies Law to appoint external directors and related Companies Law rules concerning the composition of certain board committees. Under these regulations, the exemptions from such Companies Law requirements will continue to be available to us so long as: (i) we do not have a “controlling shareholder” (as such term is defined under the Companies Law), (ii) our shares are traded on certain U.S. stock exchanges, including Nasdaq, and (iii) we comply with the director independence requirements and the audit committee and compensation committee composition requirements under U.S. laws (including applicable rules of Nasdaq) applicable to U.S. domestic issuers.
We are a “foreign private issuer” (as such term is defined in Rule 3b-4 under the Exchange Act). As a foreign private issuer, we are permitted to comply with Israeli corporate governance practices instead of the corporate governance rules of Nasdaq, provided that we disclose which Nasdaq requirements we are not following and the equivalent Israeli requirement. As a foreign private issuer and in accordance with Nasdaq Listing Rule 5615(a)(3), we may, and have elected to, comply with home country (Israel) governance requirements and certain exemptions thereunder rather than complying with certain of the corporate governance requirements of the Nasdaq.
In accordance with Israeli law and practice and subject to the exemption set forth in Rule 5615 of the Nasdaq Listing Rules, we have elected to follow the provisions of the Companies Law, rather than the Nasdaq Listing Rules, with respect to the following requirements:
| ● | Quorum. Under the corporate governance rules of Nasdaq, a quorum requires the presence, in person or by proxy, of holders of at least 33⅓% of the total issued outstanding voting power of our shares at each general meeting of shareholders, pursuant to the Amended Articles, and as permitted under the Companies Law, the quorum required for a general meeting of shareholders consists of at least two shareholders present in person or by proxy in accordance with the Companies Law, who hold or represent at least 33⅓% of the total outstanding voting power of our shares, except if (i) any such general meeting of shareholders was initiated by and convened pursuant to a resolution adopted by the board of directors and (ii) at the time of such general meeting, we qualify to use the forms and rules of a “foreign private issuer,” in which case the requisite quorum consists of two or more shareholders present in person or by proxy who hold or represent at least 25% of the total outstanding voting power of our shares (and if the meeting is adjourned for a lack of quorum, the quorum for such adjourned meeting will be, subject to certain exceptions, any number of shareholders). |
| ● | Shareholder approval. We will seek shareholder approval for all corporate actions requiring such approval under the requirements of the Companies Law, rather than pursuant to Nasdaq Listing Rule 5635. In particular, under this Nasdaq rule, shareholder approval would otherwise generally be required for: (i) an acquisition of shares/assets of another company that involves the issuance of 20% or more of the acquirer’s shares or voting rights or if a director, officer or 5% shareholder has greater than a 5% interest in the target company or the consideration to be received; (ii) the issuance of shares leading to a change of control; (iii) adoption/amendment of equity compensation arrangements; and (iv) issuances of 20% or more of the shares or voting rights (including securities convertible into, or exercisable for, equity) of a listed company via a private placement (and/or via sales by directors/officers/5% shareholders) if such equity is issued (or sold) at below the lower of: (x) the Nasdaq Official Closing Price (as reflected on Nasdaq.com) immediately preceding the signing of the applicable binding agreement; and (y) the average Nasdaq Official Closing Price (as reflected on Nasdaq.com) for the five trading days immediately preceding the signing of the applicable binding agreement. Under the Israeli Companies Law, the adoption of, and material changes to, equity-based compensation plans generally require the approval of the board of directors. |
| ITEM 16H. | MINE SAFETY DISCLOSURE |
Not applicable.
190
| ITEM 16I. | DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS |
Not applicable.
| ITEM 16J. | INSIDER TRADING POLICIES |
Not applicable.
| ITEM 16K. | CYBERSECURITY |
Cybersecurity Risk Management and Strategy
We have developed and maintain cybersecurity risk management protocols and policies intended to protect the confidentiality, integrity, and availability of our critical systems and information and to manage risks related to our network and cloud security, including security measures and controls to identify, protect and detect cybersecurity risks, and to respond to, and recover from cybersecurity incidents. We provide cybersecurity awareness training for our employees and all new employees must confirm receipt and review of our protocols and policies.
We have engaged a third-party to provide operational support for cybersecurity risks and to assess, test or otherwise assist with aspects of our security controls, including our exposure to risks from use of third party service providers, on an ongoing basis. This forms a critical part of our risk management strategy, which we believe facilitates effective management and mitigation of risks and ensures adherence to applicable regulatory and industry standards.
As of the date of this Annual Report, we do not believe that any risks from cybersecurity threats have materially affected, or are reasonably likely to materially affect, us, including our business strategy, results of operations, or financial condition. However, despite our efforts, we cannot eliminate all risks from cybersecurity threats, or provide assurances that we have not experienced an undetected cybersecurity incident. For more information about these risks, please see Item 3.D. “Risk Factors – Risks Related to Our Business and Our INVU Platform – Failure to maintain the security and functionality of our information systems, or to defend against or otherwise prevent a cybersecurity attack or data breach, could adversely affect our business, financial position, results of operations and liquidity.”
Cybersecurity Governance
Our audit committee provides oversight of our cybersecurity risk management and of our protocols and processes for managing cybersecurity risks in the context of overall risk management oversight.
Our cybersecurity risk management and the related protocols and processes are overseen by our management team, which will provide applicable updates to our audit committee regarding cybersecurity threats if and when they materialize, or upon the occurrence of any cybersecurity incident. Our management team is responsible for the supervision of our retained external cybersecurity consultants, who oversee the systems upon which we rely to identify cybersecurity incidents.
191
PART III
| ITEM 17. | FINANCIAL STATEMENTS |
The registrant has responded to Item 18 in lieu of responding to this Item 17.
| ITEM 18. | FINANCIAL STATEMENTS |
The following financial statements, the related notes thereto, and the Report of Independent Public Accountants are filed as a part of this Annual Report on Form 20-F.
192
NUVO GROUP LTD.
INDEX TO FINANCIAL STATEMENTS
F-1

Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of
Nuvo Group Ltd.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Nuvo Group Ltd. and its subsidiary (the “Company”) as of December 31, 2023 and 2022, and the related consolidated statements of income, comprehensive income, changes in shareholders’ capital deficiency and cash flows for each of the three years in the period ended December 31, 2023, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023 in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt About the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1b to the consolidated financial statements, the Company has not generated significant revenues from its operations and has suffered recurring losses from operations and negative cash flows from operations. These circumstances raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1b. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/
Certified Public Accountants (Isr.)
A member of PricewaterhouseCoopers International Limited
May 7, 2024
We have served as the Company’s auditor since 2022.
F-2
NUVO GROUP LTD.
CONSOLIDATED BALANCE SHEETS
(U.S. dollars in thousands)
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash | ||||||||
| Accounts receivable, net of credit losses of zero as of December 31, 2023 and 2022, respectively | ||||||||
| Other current assets | ||||||||
| Inventory | ||||||||
| TOTAL CURRENT ASSETS | $ | $ | ||||||
| NON-CURRENT ASSETS: | ||||||||
| Property and equipment, net | ||||||||
| Restricted cash | ||||||||
| Severance pay fund | ||||||||
| Other assets | ||||||||
| TOTAL NON-CURRENT ASSETS | ||||||||
| TOTAL ASSETS | $ | $ | ||||||
| LIABILITIES AND REDEEMABLE CROSSOVER PREFERRED SHARES, NET OF CAPITAL DEFICIENCY | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Accounts payable and accruals: | ||||||||
| Trade | ||||||||
| Other | ||||||||
| Commitment to shareholder (see Note 17) | ||||||||
| SAFE liability | ||||||||
| Convertible loans | ||||||||
| Current maturities of bridge loans | ||||||||
| TOTAL CURRENT LIABILITIES | ||||||||
| NON-CURRENT LIABILITIES | ||||||||
| Accrued severance pay | ||||||||
| Bridge loans | ||||||||
| Redeemable crossover preferred shares - put option derivative | ||||||||
| COMMITMENTS AND CONTINGENT LIABILITIES (see Note 12) | ||||||||
| TOTAL LIABILITIES | $ | $ | ||||||
| Redeemable crossover preferred shares and incentive shares, par value NIS per share; and zero shares authorized as of December 31, 2023 and 2022; and zero shares issued and outstanding at December 31, 2023 and 2022, respectively | ||||||||
| SHAREHOLDERS’ CAPITAL DEFICIENCY: | ||||||||
| Ordinary shares, par value NIS per share; shares authorized as of December 31, 2023 and 2022; shares and shares issued and outstanding at December 31, 2023 and 2022, respectively | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( |
) | ( |
) | ||||
| Total redeemable crossover preferred shares and capital deficiency | ( |
) | ( |
) | ||||
| Total liabilities, net of capital deficiency | $ | $ | ||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-3
NUVO GROUP LTD.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(U.S. dollars in thousands, except share and per share data)
| Year Ended December 31, |
||||||||||||
| 2023 | 2022 | 2021 | ||||||||||
| Revenues | $ | $ | $ | |||||||||
| Cost of revenues | ||||||||||||
| GROSS LOSS | ( |
) | ||||||||||
| Operating expenses | ||||||||||||
| Research and development, net | ||||||||||||
| Sales and marketing | ||||||||||||
| General and administrative | ||||||||||||
| Total operating expenses | ||||||||||||
| LOSS FROM OPERATIONS | ( |
) | ( |
) | ( |
) | ||||||
| Change in fair value of financial instruments | ( |
) | ( |
) | ||||||||
| Other financial expenses, net | ( |
) | ( |
) | ( |
) | ||||||
| LOSS BEFORE TAX EXPENSE | ( |
) | ( |
) | ( |
) | ||||||
| TAX EXPENSES | ( |
) | ||||||||||
| TOTAL COMPREHENSIVE LOSS | $ | ( |
) | $ | ( |
) | $ | ( |
) | |||
| NET LOSS PER SHARE - BASIC AND DILUTED | $ | ( |
) | $ | ( |
) | $ | ( |
) | |||
| WEIGHTED AVERAGE NUMBER OF SHARES USED IN COMPUTING NET LOSS PER SHARE - BASIC AND DILUTED | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-4
NUVO GROUP LTD.
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ CAPITAL DEFICIENCY
(U.S. dollars in thousands, except share data)
| Additional | ||||||||||||||||||||
| Ordinary shares | Paid-in | Accumulated | ||||||||||||||||||
| Number | Amount | capital | Deficit | Total | ||||||||||||||||
| Balance as of January 1, 2021 | $ | $ | $ | ( |
) | $ | ( |
) | ||||||||||||
| Exercise of options | * | |||||||||||||||||||
| Share-based compensation | - | |||||||||||||||||||
| Comprehensive loss | - | ( |
) | ( |
) | |||||||||||||||
| Balance as of December 31, 2021 | $ | $ | $ | ( |
) | $ | ( |
) | ||||||||||||
| Exercise of options | * | |||||||||||||||||||
| Share-based compensation | - | |||||||||||||||||||
| Comprehensive loss | - | ( |
) | ( |
) | |||||||||||||||
| Balance as of December 31, 2022 | ( |
) | ( |
) | ||||||||||||||||
| Exercise of options (see note 13d) | * | * | * | |||||||||||||||||
| Share-based compensation | - | |||||||||||||||||||
| Bridge loan warrants | - | |||||||||||||||||||
| Settlement of commitment to shareholder | - | |||||||||||||||||||
| Comprehensive loss | - | ( |
) | ( |
) | |||||||||||||||
| Balance as of December 31, 2023 | $ | $ | $ | ( |
) | $ | ( |
) | ||||||||||||
| * |
Represents an amount less than $1. |
The accompanying notes are an integral part of the consolidated financial statements.
F-5
NUVO GROUP LTD.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands)
| Year Ended December 31, |
||||||||||||
| 2023 | 2022 | 2021 | ||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net loss | $ | ( |
) | $ | ( |
) | $ | ( |
) | |||
| Adjustments to reconcile net loss to net cash used in operating activities | ||||||||||||
| Depreciation and amortization | ||||||||||||
| Remeasurement of financial instruments | ( |
) | ||||||||||
| Share-based compensation | ||||||||||||
| Non-cash interest expense | ||||||||||||
| Other financial expense (income), net | ( |
) | ||||||||||
| Changes in fair value of commitment to shareholder | ( |
) | ( |
) | ||||||||
| Amortization of debt discount | ||||||||||||
| Loss (gain) on amounts funded in respect of severance pay | ( |
) | ||||||||||
| Changes in operating assets and liabilities | ||||||||||||
| Inventory | ( |
) | ( |
) | ||||||||
| Accounts receivable, net | ( |
) | ||||||||||
| Other current assets | ( |
) | ( |
) | ( |
) | ||||||
| Other assets | ( |
) | ||||||||||
| Trade payables | ( |
) | ( |
) | ||||||||
| Other accounts payable | ( |
) | ||||||||||
| Convertible loans | ( |
) | ||||||||||
| Accrued severance pay | ( |
) | ||||||||||
| Net cash used in operating activities | ( |
) | ( |
) | ( |
) | ||||||
| Cash flows from investing activities: | ||||||||||||
| Amounts funded in respect of severance pay | ( |
) | ( |
) | ||||||||
| Purchase of property and equipment | ( |
) | ( |
) | ( |
) | ||||||
| Net cash used in investing activities | ( |
) | ( |
) | ( |
) | ||||||
| Cash flows from financing activities: | ||||||||||||
| Exercise of options | ||||||||||||
| Proceeds from issuance of convertible loans | ||||||||||||
| Proceeds from issuance of SAFE liability | ||||||||||||
| Repayment of convertible loans | ( |
) | ||||||||||
| Proceeds from redeemable crossover preferred shares | ||||||||||||
| Proceeds from bridge loans and warrants | ||||||||||||
| Net cash provided by financing activities | ||||||||||||
| Effect of exchange rate changes on cash, cash equivalents and restricted cash | ( |
) | ( |
) | ||||||||
| (Decrease) in cash and cash equivalents, and restricted cash | ( |
) | ( |
) | ||||||||
| Cash, cash equivalents and restricted cash at the beginning of the year | $ | $ | $ | |||||||||
| Cash, cash equivalents and restricted cash at the end of the year | $ | $ | $ | |||||||||
| Supplemental disclosure of cash flow information: | ||||||||||||
| Cash paid for income taxes | $ | $ | $ | |||||||||
| Cash paid for interest | $ | $ | $ | |||||||||
| Noncash investing and financing activities | ||||||||||||
| Cashless exercise of stock options | $ | $ | $ | |||||||||
| Issuance of SAFE liability | $ | $ | $ | |||||||||
| Settlement of commitment to shareholder | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-6
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
NOTE 1 - DESCRIPTION OF BUSINESS
| a. | General |
Nuvo Group Ltd. (the “Company”) was incorporated under the laws of Israel and commenced operations in June 2006.
The Company operates in one line of business and is engaged in research, development and marketing of innovative medical devices and services for pregnancy monitoring.
In 2009 the Company established a wholly-owned subsidiary under the laws of the State of Delaware, Nuvo Group USA, Inc. (the “Subsidiary”), which provides distribution services under an intercompany distribution agreement with the Company.
As of December 31, 2023 and 2022, substantially all of the Company’s long-lived assets are located in Israel.
| b. | Liquidity and Going Concern |
The Company is engaged in research and development activities, currently commercializing its product, INVU, which has not yet generated material revenues from operations. The Company has an accumulated deficit as of December 31, 2023, as well as a history of net losses and negative operating cash flows since its inception. The Company has funded its operations through financing, mainly equity issuance, convertible loans agreements (“Convertible Loans”) and Simple Agreements for Future Equity (“SAFE”). The Company expects to continue to incur losses and negative cash flows from operations until INVU reaches commercial profitability. As a result of these expected losses and negative cash flows from operations, along with the Company’s current cash position, the Company does not have sufficient resources to fund operations for the next 12 months from the issuance of these financial statements. These circumstances raise a substantial doubt about the Company’s ability to continue as a going concern. These financial statements have been prepared assuming that the Company will continue as a going concern and do not include any adjustments that might result from the outcome of this uncertainty. Management’s plans include the continued development and commercialization of the Company’s product. For this purpose, the Company intends to raise additional financing through the sale of additional equity securities, incurrence of debt, or capital inflows from strategic partnerships.
In the context of obtaining sustainable funding and in order to continue as a going concern, the Company has evaluated a broad range of financing options. On May 1, 2024, the Company completed its previously announced de-SPAC merger transaction with LAMF Global Ventures Corp I (“LAMF”), a NASDAQ listed Special Purpose Acquisition Company (“SPAC”) (see below). Following the de-SPAC, the Company still does not have sufficient resources to fund its operations for the next 12 months and will continue to depend on additional funding in the future.
There is no assurance, however, that the Company will be successful in obtaining the level of financing needed for its operations. If the Company is unsuccessful in commercializing its products and raising capital, it may be compelled to delay, restrict, reduce, or terminate its current activities or even discontinue one or more of its development programs entirely.
F-7
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
Business Combination Agreement (BCA) and the de-SPAC transaction:
On April 25, 2023 the Company and LAMF signed a non-binding letter of intent (the “LOI”), contemplating entry into the BCA and its consummation, which was extended first on May 16, 2023 and then on May 31, 2023, and expired on June 14, 2023.
On May 11, 2023, LAMF held an extraordinary general meeting of shareholders. In this meeting, LAMF Shareholders approved amendments to the existing governing documents to extend the date by which LAMF must complete an initial business combination from May 16, 2023 to November 16, 2023, and to allow LAMF, without another shareholder vote, by resolution of the LAMF Board, to elect to further extend the date by which LAMF must complete an initial business combination in one-month increments up to six additional times, or a total of up to twelve months total, up to May 16, 2024. On January 8, 2024, the LAMF Board elected to extend the extended date to February 16, 2024 through an additional monthly extension. LAMF’s articles of association provides that the Company has the right to extend the deadline date up to three additional times for an additional one month each time, from February 16, 2024, the current deadline date, to up to May 16, 2024.
On May 1, 2024, the Company completed its previously announced de-SPAC merger transaction with LAMF. Each Nuvo Share issued and outstanding will, by virtue of the Acquisition Merger and upon the terms and subject to the conditions set forth in the Business Combination Agreement, automatically be deemed to have been transferred and automatically deemed for all purposes to represent only the right to receive a number of Holdco Ordinary Shares equal to the Equity Exchange Ratio. Each Nuvo Crossover Preferred Share issued and outstanding will represent only the right to receive a number of Holdco Preferred Shares. Each Nuvo Warrant issued and outstanding will be assumed by Holdco, and each such Nuvo Warrant shall be converted into a warrant to purchase Holdco Ordinary Shares (each, a “Converted Warrant”). Each Converted Warrant shall continue to have and be subject to the same terms and conditions as were applicable to such Nuvo Warrant immediately before the Acquisition Effective Time (including expiration date and exercise provisions), except as explicitly set forth in the Business Combination Agreement. All of the outstanding and unexercised options to purchase Nuvo Shares, whether or not then vested or fully exercisable, granted to any current or former employee, officer, director or other service provider of Nuvo will, automatically be assumed by Holdco, and each such Nuvo Option shall be converted into an option to purchase Holdco Ordinary Shares (each, a “Converted Option”). Each Converted Option shall continue to have and be subject to the same terms and conditions as were applicable to such Nuvo Option except as explicitly set forth in the Business Combination Agreement. The Company began trading on the Nasdaq on May 2, 2024 as Holdco Nuvo Group DG Ltd. (NUVO).
| c. | Risks Related to Our Operations in Israel including the recent attack by Hamas and other terrorist organizations from the Gaza Strip and Israel’s war against them. |
In October 2023, Israel was attacked by a terrorist organization and entered a state of war. As of the date of these consolidated financial statements, the war in Israel is ongoing and continues to evolve. The Company operations including the production facility are located in Israel. Currently, such activities in Israel remain largely unaffected. During the year ended December 31, 2023, the impact of this war on the Company’s results of operations and financial condition was immaterial. However, at this time, it is not possible to predict the intensity or duration of the war, nor can we predict how this war will ultimately affect Israel’s economy in general, and The Company continues to monitor the situation closely and examine the potential disruptions that could adversely affect its operations.
F-8
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES:
| a. | Basis of presentation |
The accompanying audited consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”).
| b. | Use of Estimates |
The preparation of financial statements in conformity with GAAP requires management to make estimates, judgments and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. The Company evaluates on an ongoing basis its assumptions, including those related to contingencies, income tax uncertainties, share-based compensation cost, useful lives of other assets, and fair value measurement of SAFE liability, commitment to shareholder and convertible loans. The Company bases these estimates on historical and anticipated results, trends and various other assumptions that it believes are reasonable under the circumstances, including assumptions as to future events. Actual results could differ from those estimates.
| c. | Functional Currency: |
The Company’s financing rounds and financing agreements are denominated in United States dollars (“Dollars” or “U.S. dollars”). The Company’s management believes that the Dollar is the primary currency of the economic environment in which the Company operates. It is further expected that the Company’s current revenues will be denominated mainly in Dollars. Thus, the functional currency of the Company is the U.S. dollar. Accordingly, monetary accounts maintained in currencies other than the dollar are re-measured into dollars in accordance with Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 830 “Foreign Currency Matters”.
Changes in currency exchange rates between the Company’s functional currency and the currency in which a transaction is denominated are included in the Company’s statements of comprehensive loss as financial expenses, net, in the period in which the currency exchange rates change.
| d. | Principles of consolidation: |
The consolidated financial statements include the accounts of the Company and its wholly owned Subsidiary. Intercompany balances and transactions have been eliminated upon consolidation.
| e. | Cash and cash equivalents and restricted cash: |
Cash equivalents are short-term highly liquid investments that are readily convertible to cash, with original maturities of three months or less, when purchased.
Restricted cash is primarily invested in deposits, to secure obligations under the Company’s lease agreements and to secure Company-issued credit cards.
F-9
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
The following table provides a reconciliation of the cash and cash equivalents balances reported on the consolidated balance sheets and the cash, cash equivalents and restricted cash balances reported in the consolidated statements of cash flows:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash - current assets | ||||||||
| Restricted cash - long-term assets | ||||||||
| Total cash, cash equivalents, and restricted cash | $ | $ | ||||||
| f. | Accounts Receivable, Net |
Accounts receivable, net are recorded at the invoiced amount and are non-interest bearing. The Company does not have a history of credit losses related to accounts receivables. The Company applies the Current Expected Credit Losses (CECL) methodology for estimating allowances for credit losses. The estimate of expected credit losses is based on an aging schedule which utilizes relevant information about past events, current conditions, and reasonable and supportable forecasts that affect the collectability of the reported amounts. The Company had zero expected credit losses as of December 31, 2023 and 2022, respectively.
The Company receives payments from customers based on a billing schedule as established in its customer contracts. Accounts receivable are recorded when the Company has a contractual right to consideration. In some arrangements, a right to consideration for the Company’s performance under the customer contract may occur before invoicing the customer, resulting in unbilled accounts receivable.
| g. | Property and equipment, net: |
Property and equipment are stated at cost, net of accumulated depreciation.
Depreciation is calculated using the straight-line method over the estimated useful lives of the assets, at the following annual rates:
| % | ||
| Computers and software | ||
| Office furniture and equipment | ||
| Electronic equipment | ||
| Leasehold improvements |
| h. | Impairment of long-lived assets: |
The Company’s long-lived assets are reviewed for impairment in accordance with ASC 360-10-35, “Property, Plant, and Equipment- Subsequent Measurement,” whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The carrying amount of a long-lived asset (asset group) is not recoverable if it exceeds the sum of the future undiscounted cash flows expected to be generated by such assets. Impairment is recognized at the amount by which the carrying amount of the assets exceeds the fair value of the assets. In 2023 and 2022,
F-10
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| i. | Leases: |
The Company’s leases are accounted for under ASC 842, “Leases”. Operating leases are included in operating lease right-of-use (“ROU”) assets and operating lease liabilities in the balance sheet. The Company elected the short-term lease recognition exemption for leases with a lease term of 12 months or less.
ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent its obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at the commencement date based on the present value of lease payments over the lease term. The Company uses its incremental borrowing rate based on the information available at the commencement date to determine the present value of the lease payments. The Company elected the practical expedient to not separate lease and non-lease components for all of the Company leases.
The Company subsequently measures the ROU asset at the present value of the remaining lease payments, adjusted for the remaining balance of any lease incentives received, any cumulative prepaid or accrued rent if the lease payments are uneven throughout the lease term and any unamortized initial direct costs. Further, the Company will recognize lease expense on a straight-line basis over the lease term.
As of December 31, 2023 and 2022, the Company does not have any finance leases and had one short-term operating lease.
Leases with an initial term of 12 months or less that contain purchase options or renewal terms that the Company is not reasonably certain to exercise or leases with an initial term of more than 12 months that contain termination options exercisable in less than 12 months that the Company is not reasonably certain to not exercise, are not recorded on the consolidated balance sheet. The Company recognizes the lease expense for such leases on a straight-line basis in the statements of comprehensive loss over the lease term.
| j. | Severance pay: |
Israeli labor law generally requires payment of severance pay upon dismissal of an employee or upon termination of employment in certain other circumstances.
Pursuant to section 14 of the Israeli Severance Compensation Act, 1963, most of the Company’s employees are entitled to have monthly deposits, at a rate of % of their monthly salary, made in their name with insurance companies. Payments in accordance with section 14 relieve the Company from any future severance payments to these employees. The severance pay expenses for such employees were approximately $
The Company’s liability for severance pay for one of its Israeli employees is calculated pursuant to Israeli Severance Pay Law, 1963 (the “Israeli Severance Pay Law”) based on the most recent salary of the employee multiplied by the number of years of employment, as of the balance sheet date. This employee is entitled to one month’s salary for each year of employment or a portion thereof and to receive additional severance pay. The Company records the liability as if it were payable at each balance sheet date on an undiscounted basis. The liability is classified based on the expected date of settlement, and therefore is usually classified as a long-term liability, unless the cessation of the employees is expected during the upcoming year.
The Company’s liability for this Israeli employee is partially provided for by monthly deposits for insurance policies and the remainder by an accrual. The value of these policies is recorded as an asset in the Company’s balance sheet.
The deposited funds include profits and losses accumulated up to the balance sheet date. The deposited funds may be withdrawn only upon the fulfillment of the obligation pursuant to the Israeli Severance Pay Law or labor agreements. The value of the deposited funds is based on the cash redemption value of these policies.
F-11
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
In addition, the Company has deposited certain amounts with a trustee, to compensate for any severance pay liability that is not covered by other funds. These deposits are restricted and may be withdrawn only for payment of severance pay liabilities. The severance pay funds and the restricted deposits for employee benefits are classified based on the classification of the corresponding liability.
The severance pay expenses for such employees were approximately $
| k. | Other assets: |
The Company produces a wearable sensor band (“Band”) device which the Company considers an integral part of the Company’s service offering. The Bands are used numerous times and have useful lives beyond one year. Each time a Band is used over an expected lifetime of approximately three years, a portion of the cost of the Band is recorded as a cost of revenue. The Company’s estimate for the number of times the same Band can be used is based on testing in research and development, loss rates, product obsolescence, and the amount of time it takes the device to go through the manufacturing, shipping, customer shelf and patient wear time and upload process. The Company considers all finished goods and raw materials to be other assets.
As of December 31, 2023 and 2022, other assets included finished goods of $
| l. | Inventory |
Inventories are stated at the lower of cost or net realizable value. Inventory write-off is provided to cover risks arising from slow-moving items, technological obsolescence, excess inventories and discontinued products. Write-offs during the years ended December 31, 2023, 2022, and 2021 were immaterial.
Inventory items are valued using the “average price” method. The Company assesses the carrying value of its inventory for each reporting period to ensure inventory is reported at the lower of cost or net realizable value in accordance with ASC 330-10-35, “Inventory”. Charges for obsolete and slow-moving inventories are recorded based upon an analysis of specific identification of obsolete inventory items and quantification of slow-moving inventory items. These assessments consider various factors, technological obsolescence, estimated current and future market values and new product introduction. In cases when there is evidence that the anticipated utility of goods, in their disposal in the ordinary course of business, will be less than the historical cost of the inventory, the Company recognizes the difference as a current period charge to earnings and carries the inventory at the reduced cost basis until it is sold or disposed of. As of December 31, 2023 and 2022, inventory was comprised of raw material and components only.
| m. | Deferred Revenue |
Revenue is deferred when the Company has the right to invoice in advance of performance under a customer contract. The current portion of deferred revenue balances are expected to be recognized in the following 12-month period and are recognized within other current liabilities. As of December 31, 2023, the Company did not have material non-current deferred revenue.
| n. | Redeemable Crossover Preferred Shares – Put Option Derivative |
The Company has assessed under ASC 815 “Derivatives and Hedging” (“ASC 815”) that the conversion feature is not clearly and closely related to the debt host and requires bifurcation as a derivative liability, which will be recorded at fair value on a recurring basis.
F-12
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| o. | Redeemable Crossover Preferred Shares |
The Company records all redeemable crossover preferred shares at their respective fair values, net of issuance costs, on the dates of issuance. Redeemable crossover preferred shares are classified outside of shareholders’ capital deficiency on the accompanying balance sheets. Because the redemption of the redeemable crossover preferred shares is contingent upon an occurrence of certain events outside of the Company’s control, their carrying values are not remeasured to their redemption values. Subsequent adjustment of the amount presented in temporary equity is required only if the Company’s management estimates that it is probable that the instrument will become redeemable and is recognized within change in fair value of financial instruments on the accompanying consolidated statements of comprehensive loss.
| p. | Warrants to purchase Ordinary Shares: |
Warrants to purchase the Company’s ordinary shares of NIS 0.01 par value each (the “Ordinary Shares”) for a fixed number of shares and are classified as equity and, as such, are not subsequently remeasured. See also Note 10 and Note 13.
| q. | Concentrations of risk: |
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents, restricted cash and accounts receivable.
Cash and cash equivalents are invested in a major bank in Israel and the United States that exceed federally insured limits. The Company believes that the financial institutions that hold the Company’s cash are financially sound, and accordingly, that minimal credit risk exists with respect to these balances. The Company has not experienced any losses due to institutional failure or bankruptcy.
During the year ended December 31, 2023, two customers accounted for
| r. | Net loss per share attributable to Shareholders: |
The Company’s basic net loss per share is calculated by dividing net loss attributable to shareholders by the weighted-average number of shares of Ordinary Shares outstanding for the period, without consideration of potentially dilutive securities. The diluted net loss per share is calculated by giving effect to all potentially dilutive securities outstanding for the period using the treasury share method or the if-converted method based on the nature of such securities, unless the effects of potentially dilutive Ordinary Shares are anti-dilutive.
The calculation of basic and diluted loss per share includes fully vested options and warrants for the Company’s Ordinary Shares at an exercise price of USD 0.01 or NIS 0.01 per share, as the Company considers them Ordinary Shares because they are exercisable for no substantial consideration.
The Company considers its redeemable crossover preferred shares to be participating securities as a holder of a redeemable crossover preferred shares would be entitled to a dividend that would be distributed to the holders of ordinary shares, at an amount equal to the greater of (i) the sum of three times the original issue price of such share, or (ii) the amount such holder would actually receive if such redeemable crossover preferred share had been converted into ordinary shares immediately prior to such distribution event. These participating securities do not contractually require the holders of such shares to participate in the Company’s losses. Because no allocation is required under the two-class method during periods of loss to participating securities that do not have a contractual obligation to share in the losses of the Company, net loss for the periods presented was not allocated to the Company’s participating securities.
F-13
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| s. | Revenue Recognition |
The Company recognizes revenue in accordance with ASC 606, Revenue from Contracts with Customers, when a customer obtains control of promised services. The amount of revenue recognized reflects the consideration that the Company expects to be entitled to receive in exchange for these services. To achieve the core principle of this standard, the Company applies the following five steps:
| ● | Identification of the contract, or contracts, with a customer; | |
| ● | Identification of the performance obligations in the contract; | |
| ● | Determination of the transaction price; | |
| ● | Allocation of the transaction price to the performance obligations in the contract; and | |
| ● | Recognition of revenue when, or as, the Company satisfies a performance obligation. |
At contract inception, the Company assesses whether each promised good or service is distinct to identify the performance obligations in the contract. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligations.
The Company derives its revenues through commercial contracts with distributors, health systems, large private practice groups and independent women’s health practices (“the customers”). The Company has two revenue models: (1) the sales model and (2) the subscription model. Substantially all the Company’s revenue is derived from the subscription model, under which the Company provides a monitoring service for high-risk pregnancy through the Band, which is leased to healthcare providers, using Company’s cloud during the time period the expectant mother is using the service (“an episode period” which is eight weeks on average). The Band is cleaned and refurbished between each episode period and then sent to the next patient.
Under the subscription model the Band remains with the expectant mother during the episode period and is then returned to the Company and prepared for use in the next episode. The Band remains the Company’s property and responsibility, and the customer pays a fixed fee per the number of episode prescriptions.
The Company accounts for revenue earned from subscriptions, wherein an identified asset is transferred to the customer and the customer has the ability to control that asset under ASC 842. The lease of the Band under the subscription model meets the classification of an operating lease. The Company has elected to aggregate the lease and non-lease components and record the revenue combined, over the lease term. The episodes are the period over which the Company recognizes revenue, based on time elapsed. Revenue from the operating lease is generally recognized on a straight-line basis over the service period.
Under the sales model, healthcare providers purchase the Band as well as monitoring sessions, or episodes of care. Under this model, the healthcare provider owns the Band and utilizes it for monitoring sessions for its patients. Between each episode period, the Band is cleaned and refurbished and sent to the next patient. Revenue is allocated to the sale of the Band, each episode period, and each refurbishment. Revenue from the Band is recorded upon transfer of the Band to the healthcare provider, episode revenue is recorded over the eight-week period it is used, and a portion of the revenue is allocated to each refurbishment between episode periods. Generally, the Company will collect cash in advance and should therefore consider the existence of a significant financing component. However, the Company has elected to apply the practical expedient under ASC 606 which exempts the adjustment of the consideration for the existence of a significant financing component when the period between the transfer of the services and the payment for such services is one year or less.
F-14
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| t. | Cost of Revenue |
Cost of revenue primarily consists of inventory cost including materials cost, subcontracting manufacturing cost, and shipping and handling costs incurred in supporting revenue generating activities. In addition, cost of revenue includes amortization of the Bands used to fulfil the subscription services as well as royalties paid to the government in return for a government grant previously received.
| u. | Research and Development Costs, Net: |
Research and development costs are charged to the statements of comprehensive loss as incurred, net of government grants, which represent participation in research and development.
Research and development expenses include costs directly attributable to the conduct of research and development programs, including the payroll costs, lab expenses, materials, consumables, and consulting fees. All costs associated with research and development are expensed as incurred. The Company receives royalty-bearing grants, which represents participation of the Israel Innovation Authority (hereafter “IIA”) in approved programs for research and development. These grants are recognized as a reduction of research and development expenses as the related costs are incurred. In 2022 the Company received grants from the IIA and recorded $
The Company is committed to pay royalties to the Israeli Government at a rate of
| v. | Sales and Marketing |
Sales and marketing expenses primarily consist of personnel related expenses, including salaries and share-based compensation and marketing and business development expenses. The Company expenses sales and marketing as incurred.
| w. | General and Administrative |
General and administrative expenses primarily consist of personnel-related expenses associated with finance, legal, and human resources personnel, including salaries and share-based compensation expenses. In additional to personnel-related expenses, general and administrative expenses consist of rent, utilities, software expense, changes in the fair value of the commitment to shareholder, and external professional services, including accounting, audit, tax, finance, legal, compliance, and information technology. General and administrative expenses are expensed as incurred.
| x. | Fair Value of Financial Instruments: |
The Company accounts for financial instruments under ASC 820, Fair Value Measurements (“ASC 820”). This statement defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles, and expands disclosures about fair value measurements.
To increase consistency and comparability in fair value measurements, ASC 820 establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three levels as follows:
| ● | Level 1 - quoted prices (unadjusted) in active markets for identical assets or liabilities; |
F-15
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| ● | Level 2 - observable inputs other than Level 1, quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets and liabilities in markets that are not active, and model-derived prices whose inputs are observable or whose significant value drivers are observable. | |
| ● | Level 3 - assets and liabilities whose significant value drivers are unobservable. |
Observable inputs are based on market data obtained from independent sources, while unobservable inputs are based on the Company’s market assumptions.
Unobservable inputs require significant management judgment or estimation. In some cases, the inputs used to measure an asset or liability may fall into different levels of the fair value hierarchy. In those instances, the fair value measurement is required to be classified using the lowest level of input that is significant to the fair value measurement. Such determination requires significant management judgment.
Convertible Loans
During the years ended December 31, 2023 and 2022, the Company entered into certain Convertible Loans. In accordance with ASC 480, Distinguishing Liabilities from Equity (“ASC 480”), the Convertible Loans were classified as liabilities. The Company has elected the fair value option for the recognition of Convertible Loans, in accordance with ASC 825 Financial Instruments, with changes in fair value recognized in the statements of comprehensive loss. Any changes in the fair value of liabilities resulting from changes in instrument-specific credit risk are reported in other comprehensive loss and were immaterial during the years ended December 31, 2023 and 2022.
The fair value of the Convertible Loans has been estimated using the Market Approach – Guideline Public Company Method with the Hybrid method utilizing the Probability-Weighted Expected Return Method and the Option-Pricing Method.
The fair value option may be applied instrument by instrument, but it is irrevocable. Accrued interest for the Convertible Loans has been included in the change in fair value of financial instruments in the consolidated statements of comprehensive loss.
SAFE Agreements
During the years ended December 31, 2020 through 2022, the Company entered into certain SAFE agreements. In accordance with ASC 480, the Company accounts for a SAFE as a liability at fair value and adjusts the instrument to fair value at each reporting period. This liability is subject to re-measurement at each balance sheet date until a triggering event, equity financing or a liquidity or dissolution occurs, and any change in fair value is recognized in the Company’s statements of comprehensive loss. The fair value of these SAFE has been estimated using the Market Approach – Guideline Public Company Method with the Hybrid method utilizing the Probability-Weighted Expected Return Method and the Option-Pricing Method.
The carrying amounts of the Company’s other financial assets and liabilities, such as cash and cash equivalents, restricted cash, accounts receivable, and accounts payable, approximate the respective fair value due to the short-term nature of these instruments. The amounts funded in respect of employee rights are stated at cash surrender value which approximates its fair value.
F-16
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| y. | Share-Based Compensation: |
The Company accounts for share-based payments to employees and consultants, including grants of service-based employee share options in accordance with ASC 718, Compensation—Share-based Compensation, which requires that share-based payments be recognized in the statements of comprehensive loss based on their fair values. The Company accounts for forfeitures of share-based awards as they occur. The Company recognizes compensation cost for options and share awards that have a graded vesting schedule and contain only service condition on a straight-line basis for the entire award. Expense for other share-based compensation expense is recognized over the awards’ vesting period using the accelerated method.
The Company uses the Black-Scholes option-pricing model to estimate fair value of share-based awards. The Black-Scholes option-pricing model requires the use of the following assumptions:
| ● | Expected term—The expected term represents the period that the share-based awards are expected to be outstanding. For option grants that are considered to be “plain vanilla”, the expected option term was calculated based on the simplified method, which uses the midpoint between the vesting date and the contractual term, as the Company does not have sufficient historical data to develop an estimate based on participant behavior. For options granted to non-employees, the expected life of the option used is the contractual term of each such option. |
| ● | Expected volatility—Since the Company is not yet a public company and does not have any trading history for its ordinary share, the expected volatility was estimated based on the average historical volatilities of ordinary share of comparable publicly traded entities over a period equal to the expected term of the share option grants. The comparable companies were chosen based on their size, stage in the life cycle or area of specialty. The Company will continue to apply this process until enough historical information regarding the volatility of its share price becomes available. |
| ● | Risk-free interest rate—The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of grant for zero-coupon U.S. Treasury notes with maturities approximately equal to the expected term of the awards. |
| ● | Expected dividend—The Company has never paid dividends on the ordinary share and has no plans to pay dividends on the ordinary shares. Therefore, the Company used an expected dividend yield of zero. |
As the Company’s ordinary shares are not publicly traded, the fair value of the ordinary share has been determined by the Company’s Board of Directors with input from management, considering the Company’s most recently available third-party valuation of ordinary shares based on relevant valuation methodologies as outlined in the American Institute of Certified Public Accountants (“AICPA”) Practice Aid, “Valuation of Privately-Held-Company Equity Securities Issued as Compensation”. The Company also considered the amount of time between the independent third-party valuation dates and the grant. This included an evaluation of whether the subsequent valuation indicated that any significant change in valuation had occurred between the previous valuation and the grant date.
For awards with performance condition vesting features, compensation cost is recorded if it is probable that the performance condition will be achieved. If the Company originally estimated that it was not probable that the performance condition would be satisfied, compensation cost would not have been recognized. If the Company later determines that it is probable that the performance condition will be satisfied, it will recognize a cumulative catch-up adjustment to reflect the portion of the employee’s requisite service that has been provided to date and will continue to recognize compensation cost over the remaining requisite service period. The Company determined that the performance conditions as described above are not probable, and therefore no compensation cost was recognized.
F-17
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| z. | Legal Contingencies: |
From time to time, the Company or its subsidiary become involved in legal proceedings or are subject to claims arising in the ordinary course of business. Such matters are generally subject to many uncertainties and outcomes and are not predictable with assurance. The Company accrues for contingencies when the loss is probable, and it can reasonably estimate the amount of any such loss. There are no legal proceedings that are pending as of the date the financial statements are issued.
| aa. | Taxes |
The Company accounts for income taxes in accordance with ASC 740, “Income Taxes”. This codification prescribes the use of the asset and liability method whereby deferred tax asset and liability account balances are determined based on differences between financial reporting and tax bases of assets and liabilities and for carry-forward tax losses. Deferred taxes are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company provides a valuation allowance, if necessary, to reduce deferred tax assets to their estimated realizable value if it is more likely than not that some portion or all of the deferred tax asset will not be realized. As of December 31, 2023 and December 31, 2022 a full valuation allowance was provided by the Company. The Company accounts for uncertain tax positions in accordance with the provisions of ASC 740, “Income Taxes”. Accounting guidance addresses the determination of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the consolidated financial statements, under which a company may recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position should be measured based on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate settlement. Accordingly, as needed, the Company reports a liability for unrecognized tax benefits resulting from uncertain tax positions taken or expected to be taken in a tax return.
| bb. | Comprehensive income (loss) |
Comprehensive income (loss) includes no items other than net income (loss).
| cc. | Emerging Growth Company |
The Company is an emerging growth company, as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). Under the JOBS Act, emerging growth companies can delay the adoption of new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. The Company has elected to use the extended transition period for complying with new or revised accounting standards that have different effective dates for public and private companies until the earlier of the date that (i) the Company is no longer an emerging growth company or (ii) the Company affirmatively and irrevocably opts out of the extended transition period provided in the JOBS Act. However, the Company may early adopt certain accounting standards, as the JOBS Act does not preclude an emerging growth company from adopting a new or revised accounting standard earlier than the time that such standard applies to private companies to the extent early adoption is permitted.
| dd. | New Accounting Pronouncements: |
Recently Adopted Accounting Standards
In June 2016, the FASB issued ASU 2016-13, Financial Instruments—Credit Losses (“ASC 326”): Measurement of Credit Losses on Financial Instruments to introduce a new model for recognizing credit losses on financial instruments based on estimated current expected credit losses, or CECL. Under the new standard, an entity is required to estimate CECL on trade receivables at inception, based on historical information, current conditions, and reasonable and supportable forecasts. The guidance is effective for the Company for annual periods beginning after December 15, 2022, including interim periods within those fiscal years. Early application is permitted. The Company adopted ASC 326 on January 1, 2023, and there was no material impact on the Company’s consolidated balance sheet and the consolidated statements of comprehensive loss upon adoption.
F-18
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
Recently Issued Accounting Standards, Not Yet Adopted
In November 2023, the FASB issued ASU No. 2023-07 Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures. The ASU improves reportable segments disclosure requirements, primarily through enhanced disclosures about significant segment expenses. The ASU also requires a public entity that has a single reportable segment to provide all the disclosures required by the amendments and all existing segment disclosures in Topic 280. The ASU is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024. The Company does not expect the adoption of this guidance to have a material impact on the Company’s consolidated financial statement disclosures.
In December 2023, the FASB issued ASU 2023-09 “Income Taxes (Topic 740): Improvements to Income Tax Disclosures”. This guidance is intended to enhance the transparency and decision-usefulness of income tax disclosures. The amendments in ASU 2023-09 address investor requests for enhanced income tax information primarily through changes to disclosure regarding rate reconciliation and income taxes paid both in the U.S. and in foreign jurisdictions. ASU 2023-09 is effective for fiscal years beginning after December 15, 2024 on a prospective basis, with the option to apply the standard retrospectively. Early adoption is permitted. The Company is currently evaluating this guidance to determine the impact it may have on its consolidated financial statements disclosures.
NOTE 3 – REVENUE FROM CONTRACTS WITH CUSTOMERS
The following table summarizes revenue by timing of revenue recognition:
| Year Ended December 31, |
||||||||
| 2023 | ||||||||
| Amount | Percentage of Revenue | |||||||
| Sale of belts | ||||||||
| Refurbishment revenue | ||||||||
| Subscription revenue | ||||||||
| Total revenue | $ | % | ||||||
| Year Ended December 31, |
||||||||
| 2023 | ||||||||
| Amount | Percentage of Revenue | |||||||
| Israel | $ | |||||||
| United States | ||||||||
| Total revenue | $ | % | ||||||
F-19
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
Unbilled Accounts Receivable
The Company had an unbilled accounts receivable balance of $
Deferred Revenue
The current portion of deferred revenue represents amounts that are expected to be recognized within one year of the balance sheet date. As of December 31, 2023, the Company had $
The Company has elected the practical expedient not to disclose remaining performance obligations for contracts that are less than one year in length. The Company does not have any performance obligations extending beyond one year.
NOTE 4 – OTHER CURRENT ASSETS
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Government authorities1 | $ | $ | ||||||
| Advances to vendors | ||||||||
| Prepaid expenses | ||||||||
| Other | ||||||||
| Total other current assets | $ | $ | ||||||
| 1 |
NOTE 5 – PROPERTY AND EQUIPMENT, NET
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cost: | ||||||||
| Computers and software | $ | $ | ||||||
| Office, furniture and equipment | ||||||||
| Electronic equipment | ||||||||
| Property and equipment, gross | ||||||||
| Less: accumulated depreciation | ||||||||
| Property and equipment, net | $ | $ | ||||||
Depreciation expenses amounted to $
F-20
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
NOTE 6 – OPERATING AND SHORT-TERM LEASES
During 2017 the Company entered into an operating lease agreement, according to which the paid rent started August 2018. The monthly average rent expenses were approximately $
In August 2021, the Company signed a new sublease agreement to sublease parts of its office space in Tel Aviv to a third party for an annual consideration of approximately $
Currently the Company has two short term lease agreements with a monthly average rent expense of approximately $
NOTE 7 – OTHER ACCRUALS
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Employees and payroll accruals | $ | $ | ||||||
| Accrued expenses | ||||||||
| Accrued vacation and recuperation | ||||||||
| Tax liability | ||||||||
| Deferred revenues | ||||||||
| Other | ||||||||
| Total other accruals | $ | $ | ||||||
From August through October 2023, the Company signed several agreements to issue Redeemable Crossover Preferred Shares at a per share issuance price of $ for total proceeds of $, of which $
The Redeemable Crossover Preferred Shares received a liquidation preference, ranking them ahead of all other classes of Nuvo shareholders, equal to the greater of (i) the sum of three times the original issuance price for the Redeemable Crossover Preferred shares, or (ii) the amount such shareholders would actually receive if such Redeemable Crossover Preferred shares had been converted into Nuvo ordinary shares immediately prior to a distribution event; in each case, plus any dividends declared but unpaid on such share. Each Redeemable Crossover Preferred Share will be converted to Holdco Preferred Shares and is then convertible at the option of the shareholder beginning three years after the sale of the Shares. Upon conversion of any Redeemable Crossover Preferred Share, the number of Ordinary Shares issued for each Redeemable Crossover Preferred shall be equal to the greater of: (i) One Ordinary Share for each Crossover Preferred Share converted; or (ii) A number of Ordinary Shares equal to three times the original issue price of the Redeemable Crossover Preferred Shares divided by the Fair Market Value as defined by the Company’s Articles of Association.
F-21
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
Each Redeemable Crossover Preferred Share shall confer upon the holder thereof the rights, powers, restrictions, qualifications and limitations accruing to and imposed upon the holders of Ordinary Shares in the Articles of Association (except where expressly subject to different treatment).
The conversion option, which results in preferred shareholders receiving three times their initial investment in Preferred Shares, is in effect a share-settled put and therefore is considered an embedded derivative in a debt-like host. This embedded derivative is not clearly and closely related to the debt host and requires bifurcation as a derivative liability, which is recorded at fair value. For the fair value of the redeemable crossover preferred shares – put option derivative, see Note 11.
In addition, Holdco will also issue approximately $
During the year ended December 31, 2023, the Company received proceeds of $. The proceeds were received before the crossover preferred shares were issued and the Company determined that, until issuance, the amounts received represented a contingent forward to issue redeemable crossover preferred shares. The contingent forward was accounted for as a liability measured at fair value at each balance sheet date. Upon issuance of the crossover preferred shares, the contingent forward was reclassified to mezzanine equity in the Company’s consolidated balance sheet. For the Redeemable Crossover Preferred shares’ fair value, see Note 11. The Company did not have any Redeemable Crossover Preferred shares agreements in effect during the year ended December 31, 2022.
NOTE 9 - SAFE LIABILITY
The Company entered into SAFE agreements, with several existing shareholders and new investors, pursuant to which the Company issued to the investors the right to acquire certain shares in exchange for payment by the investors, subject to certain terms and conditions.
During the year ended December 31, 2020, a total of approximately $
During the year ended December 31, 2021, a total of approximately $
During the year ended December 31, 2022, a total of approximately $
During the year ended December 31, 2023, $
During the year ended December 31, 2022, $
F-22
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
After the execution of the BCA on August 17, 2023, the required majority of SAFE holders signed an amendment to their existing agreements. This adjusts the outstanding SAFEs to carry a valuation cap of $
The SAFEs contain certain conversion triggers which provide for the conversion of the investment into Ordinary Shares in the event of: (i) an Equity Financing or (ii) either a change of control transaction or an initial public offering, whichever occurs sooner, which in each case is referred to as a Liquidity Event. Upon the occurrence of a Liquidity Event, the investor will, at its discretion, receive either a cash payment or shares of the most senior series issued prior to the Liquidity Event. In the event of a conversion, the conversion price is calculated as either: (i) the price per share of the Ordinary Shares sold in connection with the Equity Financing less the Discount Rate, or (ii) the price per Ordinary Share equal to the pre-money valuation cap divided by the Company’s outstanding capitalization in effect immediately prior to the Equity Financing or Liquidity Event, calculated on an as converted and fully diluted basis, with the conversion price with respect to an Equity Financing equaling whichever calculation results in the issuance of the greater number of shares to the SAFE holder.
The SAFEs are considered liabilities pursuant to ASC 480 and were initially and subsequently measured at fair value with change in fair value recognized in statements of comprehensive loss based on the following analysis:
The SAFEs were first evaluated under ASC 480-10. Each SAFE was determined to be a freestanding financial instrument since it was entered into separately and apart from any of the Company’s other financial instruments or equity transactions. In addition, each SAFE is legally detachable and separately exercisable.
The SAFEs are liabilities pursuant to ASC 480-10-25-8 since the SAFEs embody an obligation that is indexed to an obligation to repurchase the Company’s shares as the Company may be obligated to repurchase the SAFEs in certain circumstances as stipulated in the agreements. Therefore, the SAFEs are required to be initially and subsequently measured at fair value with change in fair value recognized in the statements of comprehensive loss pursuant to ASC480-10-30-7 and ASC 480-10-35-5.
The Company did not assess the SAFEs for embedded derivatives since any recognized derivatives shall not be separated from the SAFEs pursuant to ASC 815-15-25-1(b), as the SAFEs are measured at fair value through profit or loss.
NOTE 10 – DEBT
Convertible Loans
During the year ended December 31, 2022, the Company entered into several loan agreements (“Convertible Loans”) with noteholders for a total amount of $
The noteholders also received SAFEs concurrent with the loan agreement at an amount equal to
During the year ended December 31, 2023, the Company entered into several loan agreements (“Convertible Loans”) with noteholders for a total amount of $
F-23
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
The Convertible Loans mature 12 months from the effective date and can be extended by an additional 12 months at the discretion of the Company. If the Company elects to extend the maturity date, the noteholders shall receive a one-time extension fee equal to 20% of the loan principal amount. This extension fee shall be issued in SAFE as well.
The Convertible Loans may be prepaid in whole or in part at any time.
Conversion terms:
| ● | The Convertible Loans may be converted to SAFEs (the “Convertible Loan SAFEs”) at any time. |
| ● | Upon the occurrence of a Qualified Financing which is defined as an equity investment of $ |
Key terms of the Convertible Loan SAFEs:
| ● | Equity Financing – Means, with respect to the Convertible Loan SAFEs, prior to the expiration of such SAFEs, if an equity financing occurs (including through an initial public offering or business combination with a special purpose acquisition company) representing investment proceeds in excess of $ |
| ● | Conversion Price – Means either: (1) the SAFE Price – which is the price per share equal to the pre-money valuation cap of $ |
| ● | Change of Control Event – Upon the occurrence of a change of control before the termination of the Convertible Loan SAFEs, the Purchase Amount shall automatically convert into the number of shares of the Company equal to the Purchase Amount divided by the Change of Control Conversion Price, calculated as the amount received by either the Company or its shareholders upon the change of control multiplied by the Discount Rate. |
| ● | Upon the occurrence of a Termination Event (as defined under the note) and subject to applicable law, the Company will be required to facilitate the return of the Purchase Amount to the Investor immediately following the occurrence of the Termination Event. |
The rest of the SAFE conditions are similar to the 2020 SAFEs’ terms. As such, it was determined to be freestanding financial instruments.
In August 2023 the Company repaid a total of $
During the same time, 44 of 46 remaining loan investors signed the loan consent form and, as a result, the associated loans were extended for one year. Consequently, the associated extension incentive of $
The Company has elected to account for the Convertible Loans using the fair value option. Refer to Note 11 for changes in fair value.
F-24
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
Bridge Loans and Warrants
During the year ended December 31, 2023, the Company entered into an agreement with a Lender (“the Lender”) to obtain financing in the amount of $
In connection with the agreement, the Company entered into a secured promissory note agreement (the “Promissory Note”) with the Lender for $
As of December 31, 2023, the Company had obtained $
In connection with the Bridge Loans, the Lender and the third-party investors received warrants to purchase a number of ordinary shares of the Company equal to twice the principal amount of their respective loan divided by a per share price of $. The warrants will be exercisable at an exercise price of NIS 0.01 at any time after issuance. The agreement provides for a total of
The Bridge Loans are subsequently accounted for under ASC 470: Debt at amortized cost using the effective interest method. The Bridge Loans contain an embedded derivative related to a put option which requires acceleration of repayment upon an event of default as defined in the loan agreement, the value of which is negligible as of December 31, 2023.
As of December 31, 2023, the future maturities of debt outstanding, including interest, are as follows:
| Fiscal years ended December 31, | Amount | |||
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| Thereafter | ||||
| Total debt outstanding | $ | |||
F-25
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
NOTE 11 – FAIR VALUE MEASUREMENT
The fair value of the Convertible Loans, SAFEs, and Commitment to Shareholder may change significantly as additional data is obtained, impacting the Company’s assumptions used to estimate the fair value of the liabilities. In evaluating this information, considerable judgment is required to interpret the data used to develop the assumptions and estimates. The estimates of fair value may not be indicative of the amounts that could be realized in a current market exchange. Accordingly, the use of different market assumptions and/or different valuation techniques may have a material effect on the estimated fair value amounts, and such changes could materially impact the Company’s results of operations in future periods.
The following table presents changes in Level 3 liabilities measured at fair value for the years ended December 31, 2023 and 2022. Unobservable inputs were used to determine the fair value of positions that the Company has classified within the Level 3 category:
| Convertible Loans |
SAFE Liability |
|||||||
| Balance as of January 1, 2022 | $ | $ | ||||||
| Issuance consideration | ||||||||
| Changes in fair value | ( |
) | ||||||
| Balance as of December 31, 2022 | $ | $ | ||||||
| Issuance consideration | ||||||||
| Repayment of principal and accrued interest | ( |
) | ||||||
| Changes in fair value | ( |
) | ||||||
| Balance as of December 31, 2023 | $ | $ | ||||||
Refer to Note 17 for information related to the commitment to shareholder that was settled during the year ended December 31, 2023.
Significant Inputs
A summary of significant inputs (Level 3 inputs) used in measuring the Convertible Loans, the SAFEs and the contingent forward to issue Redeemable Crossover Preferred shares as of December 31, 2023 is as follows:
| de-SPAC | Staying | |||||||
| Transactions | Private | |||||||
| Valuation | Valuations | |||||||
| Key assumptions: | % | % | ||||||
| Probability weighting | ||||||||
| Time to liquidity (in years) | ||||||||
| Expected volatility | % | % | ||||||
| Risk-free interest rate | % | % | ||||||
| Expected dividend yield | ||||||||
| Equity value (in thousands) | $ | $ | ||||||
F-26
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
A summary of significant inputs (Level 3 inputs) used in measuring the Convertible Loans and SAFEs as of December 31, 2022, is as follows:
| Equity | Liquidity | |||||||
| Financing | Event | |||||||
| Scenario | Scenario | |||||||
| Key assumptions: | ||||||||
| Probability weighting | % | % | ||||||
| Time to liquidity (in years) | ||||||||
| Expected volatility | % | % | ||||||
| Risk-free interest rate | % | % | ||||||
| Expected dividend yield | % | % | ||||||
| Equity value (in thousands) | $ | $ | ||||||
Contingent Forwards Redeemable Crossover Preferred Shares
The contingent forward contract to issue redeemable crossover preferred shares, incentive shares, and the redeemable crossover preferred shares – put option, was revalued from $
The fair value of the contingent forward was determined using level 3 fair value measurement inputs. A summary of the allocation of fair value to the individual components is presented below. The corresponding loss of $
| SPAC | Staying | Weighted | ||||||||||
| Transaction | Private | Average | ||||||||||
| Scenario | Scenario | Value | ||||||||||
| Probability | % | % | ||||||||||
| Incentive shares | $ | $ | $ | |||||||||
| Redeemable crossover preferred shares | ||||||||||||
| Redeemable crossover preferred - put option | ||||||||||||
| Fair value of redeemable crossover preferred shares | $ | $ | $ | |||||||||
Warrants
During the year ended December 31, 2023, the Company issued equity-classified warrants in connection with their Bridge Loans. Upon issuance, the warrants were measured at the amount allocated to them based on relative fair value, which was determined utilizing the Black-Scholes model (“BSM”). For additional information, refer to Note 10.
A summary of significant inputs (Level 3 inputs) used in measuring the non-recurring warrants during the year ended December 31, 2023, is as follows:
| December 31, 2023 | ||
| Exercise price | NIS | |
| Expected term (in years) | - | |
| Current price of the underlying share | $ | |
| Expected volatility of the underlying share | % - % | |
| Expected dividend yield on the underlying share | % | |
| Risk-free interest rate | % - % |
F-27
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
NOTE 12 - COMMITMENTS AND CONTINGENT LIABILITIES
Royalties to IIA:
Under the Company’s research and development agreements with the IIA and pursuant to applicable laws, the Company is required to pay royalties at the rate of
NOTE 13 - SHAREHOLDERS’ CAPITAL DEFICIENCY
| a. | Ordinary Shares rights: |
Each Ordinary Share confers on its holder the rights to receive notice of, and to participate and vote in, all meetings of the shareholders, to receive dividends, and to participate in the distribution of the surplus assets and funds of the Company in the event of the liquidation, dissolution or winding up of the Company, all, as set forth in the Company’s Articles of Association and subject to applicable law.
| b. | Issuance of shares: |
During the year ended December 31, 2022, four of the Company’s consultants had exercised their options to purchase Ordinary Shares in consideration of $. No options were exercised by consultants during the year ended December 31, 2023.
| c. | Warrants: |
On May 20, 2015, the Company granted
| d. | Share-based Compensation: |
On December 2015, the Board of Directors of the Company adopted the 2015 Share Incentive Plan (the “Plan”), which provides for the grant of up to options to purchase Ordinary Shares of the Company to employees, officers, directors and consultants of the Company. During 2022 the pool of options to purchase Ordinary Shares under the Plan was increased to .
Options granted under the Plan generally expire 10 years from the date of grant.
The options generally vest 25% on the first anniversary of the vesting start date and 6.25% at the end of each subsequent quarter over the course of the following three years.
On April 28, 2021, the Board of Directors of the Company adopted amendments to the Plan. The main amendment was a cashless exercise mechanism.
F-28
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
The fair value of options granted under the stock option plan during the year ended December 31, 2023 and 2022 was estimated at the date of grant using the Black-Scholes option pricing model and the following assumptions for grants:
| December 31, | ||||
| 2023 | 2022 | |||
| Risk-free interest rates | – | % | ||
| Expected life of options | – years | years | ||
| Expected volatility | – % | % | ||
| Expected dividend yield | % | % | ||
The following table summarizes the allocation of total share-based compensation expense in the statements of comprehensive loss:
| Years Ended December 31, |
||||||||||||
| 2023 | 2022 | 2021 | ||||||||||
| Research and development, net | $ | |||||||||||
| Sales and marketing | ||||||||||||
| General and administrative | ||||||||||||
| Total share-based compensation expense | $ | $ | $ | |||||||||
The following table summarizes stock option activity:
| Number of Options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
| Balance at January 1, 2023 | $ | $ | ||||||||||||||
| Granted | ||||||||||||||||
| Forfeit | ( |
) | ||||||||||||||
| Expired | ( |
) | ||||||||||||||
| Exercised | ( |
) | $ | |||||||||||||
| Balance at December 31, 2023 | $ | |||||||||||||||
| Vested and expected to vest at December 31, 2022 | $ | $ | ||||||||||||||
| Vested and expected to vest at December 31, 2023 | $ | $ | ||||||||||||||
| Exercisable at December 31, 2022 | $ | $ | ||||||||||||||
| Exercisable at December 31, 2023 | $ | $ | ||||||||||||||
The weighted-average grant date fair value of options granted during the years ended December 31, 2023 and 2022 was $ and $, respectively. The intrinsic value of options exercised for the years ended December 31, 2023 and 2022 was $ and $, respectively. The aggregate grant-date fair value of options that vested during the years ended December 31, 2023 and 2022 was $ and $, respectively. Cash received from option exercises for the years ended December 31, 2023 and 2022 was zero and $
F-29
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
For the years ended December 31, 2023, 2022, and 2021, the Company recognized $, $, and $ of share-based compensation expense relating to stock options, respectively. As of December 31, 2023, there was $
The Company granted to several directors under the plan a total of and options during the years ending December 31, 2023, and 2022 respectively.
| e. | Performance and market-based compensation: |
The founder, who is a current Board member and former Chief Innovation Officer (“former CIO”) is entitled to future option allocations according to his employment contract. The allocation of a maximum total of options with a strike price of $ will depend on the increase of the Company value achieved via capital raises or a potential sale of the Company, out of which options were granted during 2017. These options will be granted fully vested upon reaching related valuation milestones. The right to this allocation terminates 18 months after the termination of the employment in July 2024. No further options were allocated under these rights in 2023 and 2022. No other executive officers or Board members of the Company are entitled to such or similar allocations. The Company considers these allocation conditions as both market and performance conditions. They are considered market conditions since the employment contract links the option allocation to market prices for the Company’s equity. The Company also considers these allocations under performance conditions since the options’ vesting was dependent on achievement of the required valuation milestones. The associated liability was accrued as a commitment to shareholder on the Company’s consolidated balance sheets, with associated expense and change in fair value recognized within general and administrative within the statements of comprehensive loss.
Refer to note 17 for additional information.
NOTE 14 - RESEARCH AND DEVELOPMENT, NET
| Years Ended December 31, |
||||||||||||
| 2023 | 2022 | 2021 | ||||||||||
| Salaries and wages | $ | $ | $ | |||||||||
| Share-based compensation | ||||||||||||
| Rent, office and utilities, software licenses and communication | ||||||||||||
| Professional services | ||||||||||||
| Other | ||||||||||||
| Research and development, gross | $ | $ | $ | |||||||||
| Less - participation of R&D expenses (see note 2u)1 | ( |
) | ||||||||||
| Research and development, net | $ | $ | $ | |||||||||
| 1 |
F-30
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
NOTE 15 - TAXES ON INCOME
| a. | Tax laws applicable to the Company and the Subsidiary: |
Nuvo Group Ltd. is taxed under the Israeli income tax laws. The Israeli corporate income tax rate was
| b. | Carry forward tax losses: |
Nuvo Group Ltd. has accumulated losses for tax purposes in Israel of approximately $
As of December 31, 2023, the U.S. carryforward losses were $
| c. | Tax assessments: |
Tax assessments filed by the Company in Israel through the year ended on December 31, 2017 are considered to be final and tax assessments filed by the Subsidiary in the United States through the year ended on December 31, 2019 are considered to be final.
| d. | Deferred taxes: |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. As of December 31, 2023 and 2022, the Company has provided a full valuation allowance in respect of deferred income tax assets. Management currently believes that it is more likely than not that the deferred income taxes regarding the carry forward tax losses and regarding other temporary differences will not be realized in the foreseeable future.
Significant components of the Company’s deferred income tax assets are as follows:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Carry forward tax losses | $ | $ | ||||||
| Research and development expenses, net | ||||||||
| Convertible loans | ||||||||
| Other temporary differences | ||||||||
| $ | $ | |||||||
| Less - valuation allowance | ( |
) | ( |
) | ||||
| $ | $ | |||||||
| Valuation allowance | ||||||||
| Balance at beginning of year | $ | $ | ||||||
| Losses during the year | ||||||||
| Balance at end of year | $ | $ | ||||||
F-31
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
| e. | Current taxes on income |
The main reconciling item between the statutory tax rate of the Company and the effective tax rate is the change in valuation allowance in respect of tax benefits from carried forward tax losses due to uncertainty of the realization of such tax benefits.
The changes in the valuation allowance for the years ended December 31, 2023, 2022, and 2021 were as follows:
| December 31, | ||||||||||||
| 2023 | 2022 | 2021 | ||||||||||
| Balance at the beginning of the year | $ | $ | $ | |||||||||
| Changes during the year: | ||||||||||||
| Losses during the year (including foreign exchange rate effect) | ||||||||||||
| Balance at the end of the year | $ | $ | $ | |||||||||
| f. | Accounting for Uncertain Tax position |
The following is a reconciliation of the total amounts of the Company’s uncertain tax positions during the year ended December 31, 2023 and 2022:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Balance at the beginning of the year | $ | $ | ||||||
| Increase in uncertain tax position because of tax positions taken during the year: | ( |
) | ||||||
| Balance at the end of the year | $ | $ | ||||||
Tax years as early as 2020 remain open and are subject to examination in the Company’s principal tax jurisdictions. The Company does not expect a significant change to its net unrecognized tax benefits over the next 12 months.
The following table sets forth the computation of basic and diluted net loss per share attributable to Shareholders for the periods presented:
| Year Ended December 31, |
||||||||||||
| 2023 | 2022 | 2021 | ||||||||||
| Numerator: | ||||||||||||
| Net loss | $ | ( |
) | $ | ( |
) | $ | ( |
) | |||
| Denominator: | ||||||||||||
| Weighted-average ordinary shares outstanding used in computing net loss per share attributable to shareholders | ||||||||||||
| Weighted average fully vested options and warrants for the Company’s Ordinary Shares at an exercise price of NIS 0.01 per share | ||||||||||||
| Weighted average number of shares | ||||||||||||
| Net loss per share attributable to shareholders, basic and diluted | $ | ( |
) | $ | ( |
) | $ | ( |
) | |||
F-32
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
The potentially dilutive options to purchase Ordinary Shares that were excluded from the computation amounted to , , and options for the years ended December 31, 2023, 2022, and 2021, respectively, as their effect is anti-dilutive. As a result, diluted net loss per share is the same as basic net loss per share for each of the periods presented.
In addition, the Company has not considered the effect of the potential conversion of the SAFEs (see Note 9), Convertible Loans, Bridge Loans (see Note 10), or Redeemable Crossover Preferred Shares Liability (see Note 8) to Ordinary Shares of the Company in the calculation of diluted net loss per share since the conversion of these instruments is contingent upon the occurrence of future events.
NOTE 17 - RELATED PARTIES
Related party balances within the consolidated balance sheets as of December 31, 2023 and 2022 were as follows:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| SAFE liability due to related parties | $ | $ | ||||||
| Convertible loans due to related parties | $ | $ | ||||||
| Crossover preferred shares and incentives shares issued to related parties | $ | $ | ||||||
| Bridge loans due to related parties | $ | $ | ||||||
Interest expense due to related parties in connection with the convertible loans during the years ended December 31, 2023, 2022, and 2021 was $
During the year ended December 31, 2023, the Company issued
Cross-over Preferred Shares
During the year ended December 31, 2023, the Company received several investments from related parties. These investments totaled $
Related Party Expenses
In relation to a service agreement with a related party to provide project-based work services, during the years ended December 31, 2023 and 2022, the Company received such services for total consideration of $
In relation to a service agreement with a related party to provide advisory services, during the year ended December 31, 2023, the company received such services for total consideration of $
F-33
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
In July 2023, the Company entered into a consulting services agreement (the “Consulting Agreement”) with an entity wholly owned and controlled by the Company’s Interim Chief Financial Officer as of August 2023 (“the Consulting Company”), to provide consulting services to the Company. Pursuant to the Consulting Agreement, the Company will pay the Consulting Company an annual retainer of $
Commitment to Shareholder
On November 17, 2021, the Company entered into an agreement with its former CIO pursuant to which, the Company, subject to certain conditions, agreed to issue to the former CIO options to purchase
On May 29, 2023, the Company entered into an employment termination agreement (the “Termination Agreement”) with its former CIO effective January 31, 2023:
| 1. | As per the Termination Agreement, the Company agreed to pay the former CIO the following: |
| A. | 12 monthly payments, equivalent to approximately $ |
| B. | Severance payments in respect of all employment’s periods, by no later than December 31, 2023, totaling of approximately $ |
| C. | All other amounts that have not been distributed, as of the Termination Agreement date, to his pension funds during the term of his employment in a total amount of approximately $ |
| 2. |
In August 2023, following the BCA and the Redeemable Crossover Preferred shares agreements all the conditions set forth in the Termination Agreement, between the Company and the former CIO related to an option allocation in conjunction with waiving a tax liability, were met and in September 2023, options were granted to the former CIO as a result. When the options were granted, the Company de-recognized the commitment to shareholder liability and recognized the gain on extinguishment which is recorded within general and administrative expenses on the consolidated statements of comprehensive loss. |
F-34
NUVO GROUP LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars in thousands, except share data)
NOTE 18 - SUBSEQUENT EVENTS
The Company has evaluated subsequent events from the consolidated balance sheet date through May 7, 2024, the date at which consolidated financial statements were available to be issued.
In January 2024, the Company recorded $
In February 2024, the Chief Executive Officer (“CEO”) Kelly Londy resigned after accepting a leading position at a non-competitive multinational healthcare company. Following the departure, Board member Rice Powell assumed the position of CEO and Kelly Londy joined the Company’s Strategic Advisory Council.
In February through April of 2024, the Company obtained additional financing through the Bridge Loan program as described in Note 10, in the amount of $
In March 2024, the Board of Directors of the Company approved a stock option repricing (the “Option Repricing”), which was effective on March 26, 2024, (the “Repricing Date”). The Option Repricing applied to outstanding options to purchase ordinary shares of the Company that, as of the Repricing Date, are held by employees of the Company and had an exercise price per share greater than $. As of the Repricing Date, outstanding options were repriced such that the exercise price per share was reduced to $ effective as of March 26, 2024.
In March 2024, the Board of Directors of the Company granted new hire options to the Company’s CEO and new hire options to the Company’s CFO. The options vest over a period of two to four years.
On May 1, 2024, the Company completed its previously announced de-SPAC merger transaction with LAMF. Each Nuvo Share issued and outstanding will, by virtue of the Acquisition Merger and upon the terms and subject to the conditions set forth in the Business Combination Agreement, automatically be deemed to have been transferred and automatically deemed for all purposes to represent only the right to receive a number of Holdco Ordinary Shares equal to the Equity Exchange Ratio. Refer to Note 1 for additional information.
F-35
Holdco Nuvo Group D.G Ltd.
| Report of Independent Registered Public Accounting Firm | F-37 | |
| Consolidated Balance Sheets | F-38 | |
| Notes to the Consolidated Financial Statements | F-39 |
F-36
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholder of
Holdco Nuvo Group D.G Ltd.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Holdco Nuvo Group D.G Ltd and its subsidiaries (the “Company”) as of December 31, 2023 and July 20, 2023, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and July 20, 2023 in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Kesselman&Kesselman
Certified Public Accountants (Isr.)
A member of PricewaterhouseCoopers International Limited
Tel-Aviv, Israel
February 9, 2024
We have served as the Company’s auditor since 2023.
F-37
HOLDCO NUVO GROUP D.G LTD.
CONSOLIDATED BALANCE SHEETS
(U.S. dollars)
| Dec. 31, 2023 |
July 20, 2023 |
|||||||
| ASSETS | ||||||||
| Current assets | $ | $ | ||||||
| TOTAL ASSETS | $ | $ | ||||||
| LIABILITIES AND EQUITY | ||||||||
| Current liabilities – related parties | $ | $ | ||||||
| TOTAL LIABILITIES | $ | $ | ||||||
| SHAREHOLDERS’ EQUITY: | ||||||||
| Ordinary shares, no par value per share; shares authorized as of December 31, 2023 and July 20, 2023; shares issued and outstanding at December 31, 2023 and July 20, 2023 | ||||||||
| Total equity | $ | $ | ||||||
| Total liabilities and equity | $ | $ | ||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-38
HOLDCO NUVO GROUP D.G LTD.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(U.S. dollars, except share data)
NOTE 1 - SIGNIFICANT ACCOUNTING POLICIES
General Information
Holdco Nuvo Group D.G Ltd (“the Company”) was incorporated as a limited liability company under the laws of the State of Israel on July 20, 2023 for the sole purpose of effectuating the transactions described in the business combination agreement, dated August 17, 2023 (the “Business Combination Agreement”) by and among the Company, LAMF Global Ventures Corp. I, a Cayman Islands exempted company (“LAMF”), Nuvo Group Ltd., a limited liability company organized under the laws of the State of Israel (“Nuvo”), Nuvo Assetco Corp., a Cayman Islands exempted company and a wholly owned subsidiary of the Company (“Assetco”), and H.F.N Insight Merger Company Ltd., a limited liability company organized under the laws of the State of Israel and a wholly owned subsidiary of LAMF (“Merger Sub”) (the “Business Combination”).
Pursuant to the Business Combination Agreement, one day prior to the closing of the Business Combination, LAMF will be merged with and into Assetco (the “SPAC Merger”) and Assetco will continue as the surviving corporation (Assetco, in its capacity as the surviving entity of the SPAC Merger, the “SPAC Surviving Company”). On the date of the closing of the Business Combination (the “Closing”), Merger Sub will be merged with and into Nuvo (the “Acquisition Merger”) and Nuvo will continue as the surviving corporation (Nuvo, in its capacity as the surviving entity of the Acquisition Merger, the “Acquisition Surviving Sub”).
Following the consummation of the transactions contemplated by the Business Combination Agreement, the Company will be the surviving publicly-traded corporation. In addition, following the consummation of such transactions, the SPAC Surviving Company shall distribute any remaining funds in the trust account to Holdco and shall be liquidated. However, the consummation of the transactions contemplated by the Business Combination Agreement is subject to numerous conditions, and there can be no assurances that such conditions will be satisfied.
Basis of accounting
The consolidated balance sheets have been prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”). Separate statements of changes in equity, operations, comprehensive income, and cash flows have not been presented in the consolidated Financial Statements because there have been
Prepaid expenses - Transaction costs in an offering of equity securities
In the event of offering of equity securities, incremental costs that otherwise would not have been incurred are deferred and capitalized in the balance sheet as a financial asset at amortized cost instead of expensed as incurred. When cash is received from investors as part of the offering, such deferred incremental costs are derecognised and deducted from additional paid-in capital.
F-39
Note 2 - Related party transaction with Nuvo Group Ltd
During the period July 20, 2023 to December 31, 2023, Holdco Nuvo Group D.G Ltd incurred $
Note 3 - Share capital
The Company was incorporated on July 20, 2023 and issued ordinary shares for no capital contribution.
As of December 31, 2023, the Company’s authorized share capital consists of no par value ordinary shares. Each ordinary share maintains one voting right. The outstanding shares are held by a related party, an investor, former board member and interim CFO of Nuvo Group Ltd.
Note 4 - Subsequent events
Management has evaluated events subsequent to December 31, 2023 and through February 9, 2024, the date these consolidated Financial Statements were authorized for issuance by the Board of Directors. There were no events which occurred subsequent to December 31, 2023 that merited disclosure in these consolidated Financial Statements.
F-40
| ITEM 19. | EXHIBITS |
EXHIBIT INDEX
| # | Indicates management contract or compensatory plan or arrangement. | |
| † | Certain of the exhibits and schedules to this Exhibit have been omitted in accordance with Regulation S-K Item 601(a)(5). The Registrant agrees to furnish a copy of all omitted exhibits and schedules to the SEC upon its request. | |
| + | Pursuant to Item 601(b)(10)(iv) of Regulation S-K, portions of this exhibit have been omitted because the Company customarily and actually treats the omitted portions as private or confidential, and such portions are not material and would likely cause competitive harm to the Company if publicly disclosed. The Company will supplementally provide a copy of an unredacted copy of this exhibit to the U.S. Securities and Exchange Commission or its staff upon request. | |
| ++ | Furnished herewith. | |
| * | Previously filed. | |
| ** | Filed herewith. |
193
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this report on its behalf.
| Holdco Nuvo Group D.G Ltd. | ||
| May 15, 2024 | By: | /s/ Robert Powell |
| Name: | Robert Powell | |
| Title: | Chief Executive Officer and Director | |
194