
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 20-F
OR
the fiscal year ended December 31 , 2023
Commission file number 001-40277 Olink Holding AB (publ)
(Exact name of Registrant as specified in its charter)
Not Applicable
(Translation of Registrant’s name into English)
(Jurisdiction of incorporation or organization)
(Address of principal executive offices)
With copies to:
Attn: Linda Ramirez-Eaves, General Counsel
SE-753 30
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person) Securities registered or to be registered pursuant to Section 12(b) of the Act.
| Title of each class | Trading symbol(s) | Name of each exchange on which registered | ||||||
The Nasdaq Global Market* | ||||||||
* Not for trading, but only in connection with the registration of the American Depositary Shares. | ||||||||
Securities registered or to be registered pursuant to Section 12(g) of the Act.
(Title of Class)
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.
(Title of Class)
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
As of December 31, 2023, 124,342,715 common shares were outstanding, including common shares represented by American Depositary Shares.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
☒ Yes ☐ No
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. ☐Yes ☒ No
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒Yes ☐No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
☒Yes ☐No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer, "accelerated filer,” and "emerging growth company" in Rule 12b-2 of the Exchange Act.
☒ | ☐Accelerated filer | ☐Non-accelerated filer | ||||||
☐ | ||||||||
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C.
7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
☐U.S. GAAP
☒ International Financial Reporting Standards as issued by the International Accounting Standards Board
☐Other
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes ☒No
Contents
/3
ITEM 16J. INSIDER TRADING POLICIES | ||||||||
F-1 | ||||||||
/4
Special note regarding forward-looking statements
This Annual Report contains express or implied "forward-looking statements," as defined under the Private Securities Litigation Reform Act of 1995, that involve substantial risks and uncertainties. In some cases, you can identify forward- looking statements by the words “may,” “might,” “will,” “could,” “would,” “should,” “expect,” “intend,” "seek," “plan,” "outlook," “objective,” “anticipate,” “believe,” “estimate,” “predict,” "project," “potential,” “continue,” "currently," “ongoing,” or the negative of these terms, or other comparable terminology intended to identify statements about the future. You should not place undue reliance on these statements because they involve known and unknown risks, uncertainties and other important factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. The forward-looking statements and opinions contained in this Annual Report are based on our management’s beliefs and assumptions and are based upon information currently available to our management as of the date of this Annual Report and, while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. We undertake no obligation to publicly update or revise any forward-looking statements as a result of new information, future events or otherwise. The forward-looking statements contained in this Annual Report should be read in conjunction with, and are subject to and qualified by, the risks described in the "Risk Factors" section of this Annual Report, and in the company's other filings with the SEC. Forward-looking statements contained in this Annual Report include, but are not limited to, information about:
•the proposed acquisition and tender offer of Olink by Thermo Fisher (the “Offer”);
•the ability of the parties to satisfy the closing conditions of the Offer on a timely basis, if at all;
•the possibility of regulatory approvals required for the Offer not being timely obtained, if obtained at all, or being obtained subject to conditions;
•uncertainties as to how many of Olink’s shareholders will tender their shares in the offer;
•the possibility that competing offers will be made;
•the occurrence of events that may give rise to a right of one or both of Thermo Fisher and Olink to terminate the Purchase Agreement;
•negative effects of the announcement of the Offer on the market price of Olink’s common stock;
•prior to the completion of the Offer, Olink’s business experiencing disruptions due to uncertainty or other factors related to the Offer making it more difficult to maintain relationships with employees, customers, licensees, other business partners or governmental entities;
•difficulty retaining key employees;
•the outcome of any legal proceedings related to the Offer;
•the parties being unable to successfully implement integration strategies or to achieve expected synergies and operating efficiencies within the expected timeframe for completing the Offer;
•estimates of our addressable market, market growth, future revenue, key performance indicators, expenses, capital requirements and our needs for additional financing;
•our ability to successfully implement our commercial plans, including the development, launch and scaling of our Explore product line and Olink Signature platform as well as our new product Olink Flex and our new Olink Insight online platform;
•the implementation of our business model and strategic plans for our business, products and services;
•our plan to grow our library of protein biomarker targets;
•our expectations regarding the rate and degree of market acceptance of our product lines;
•our dependence on levels of research and development spending by academic and governmental research institutions and biopharmaceutical companies, a reduction in which could limit demand for our products;
•the impact of our products and our proprietary technology, Proximity Extension Assay, on the field of proteomics and the size and growth of the addressable proteomics market;
•our competitive position, and developments and projections relating to our competitors and our industry, including estimates of the size and growth potential of the markets for our products;
•the timing, scope or likelihood of domestic and foreign regulatory filings and approvals;
•our risks related to handling of hazardous materials and other regulations governing environmental safety;
•our ability to manage and grow our business and commercialize our product lines;
•our ability to develop and commercialize new products;
•the performance of third-party manufacturers and suppliers;
•our intellectual property position, including the scope of protection we are able to establish and maintain for intellectual property rights, the validity of intellectual property rights held by third parties, and our ability not to infringe, misappropriate or otherwise violate any third-party intellectual property rights;
•occurrence of cyber incidents or failure by us or our third-party service providers to maintain cybersecurity;
•the potential effects of government regulation;
/5
•our ability to retain the continued service of our key professionals and to identify, hire and retain additional qualified professionals, including sales and marketing personnel;
•our ability to obtain additional financing in future offerings, including among others, impacts of the current volatility in the global capital and credit markets and the effects of increased inflation on the cost of capital;
•occurrence of cyber incidents or failure by us or our third-party service providers to maintain cybersecurity;
•the quarterly progression of our business and major financial metrics as they relate to the seasonal nature of our customers' buying patterns;
•the impact of local, regional, and national and international economic conditions and events, including among others, rising inflation, currency exchange rates, the ongoing military conflict between Russia and Ukraine, and between Israel and Hamas, a militia and political group operating in the Gaza Strip, and developments in China;
•our ability to maintain an effective system of internal control over financial reporting and our ability to remediate any identified material weaknesses in our internal controls; and
•any lingering impacts from the COVID-19 pandemic on our business.
PART I
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS
Not applicable.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
ITEM 3. KEY INFORMATION
A.[Reserved]
B.Capitalization and Indebtedness
Not applicable.
C.Reasons for the Offer and Use of Proceeds
Not applicable.
D.Risk Factors
The following risks relate specifically to our business and should be considered carefully. Any of the risks described below or elsewhere in this Annual Report or our other filings with the SEC could have a material impact on our business, prospects, financial condition or results of operations. The risks listed below are not the only risks that we face. Additional risks unknown to us or that we currently believe are insignificant may also affect our business. As a result, the trading price of our ordinary shares and our American Depositary Shares, or ADSs, could decline and the holders could lose part or all of their investment.
The conditions under the Purchase Agreement to our consummation of the Offer may not be satisfied at all or in the anticipated timeframe.
On October 17, 2023, the Company entered into a Purchase Agreement (the “Purchase Agreement”) with Thermo Fisher Scientific Inc., a Delaware corporation (“Thermo Fisher” or “Buyer”). Under the terms of the Purchase Agreement, the consummation of the Offer is subject to customary conditions. Satisfaction of certain of the conditions is not within our control, and difficulties in otherwise satisfying the conditions may prevent, delay or otherwise materially adversely affect the consummation of the Offer. It also is possible that an event, occurrence, revelation or development of a state of circumstances or facts since the date of the Purchase Agreement may have or reasonably be expected to have a material adverse effect (as defined in the Purchase Agreement) on the Company, the non-occurrence of which is a condition to the consummation of the Offer. We cannot predict with certainty whether and when any of the required conditions will be satisfied. If the Offer does not
/6
receive, or timely receive, the required regulatory approvals and clearances, or if another event occurs delaying or preventing the Offer, such delay or failure to complete the Offer may create uncertainty or otherwise have negative consequences that may materially and adversely affect our sales, financial condition and results of operations, as well as the price per share for our common stock.
While the Offer is pending, we are subject to business uncertainties and contractual restrictions that could disrupt our business.
Whether or not the Offer is consummated, the Offer may disrupt our current plans and operations, which could have an adverse effect on our business and financial results. The pendency of the Offer may also divert management’s attention and our resources from ongoing business and operations and our employees and other key personnel may have uncertainties about the effect of the pending Offer, and the uncertainties may impact our ability to retain, recruit and hire key personnel while the Offer is pending or if it fails to close. We may incur unexpected costs, charges or expenses resulting from the Offer.
The preparations for integration between Thermo Fisher and the Company have placed, and we expect will continue to place, a significant burden on many of our personnel and on our internal resources. If, despite our efforts, key personnel depart because of these uncertainties and burdens, or because they do not wish to remain with the combined company, our business and results of operations may be adversely affected. In addition, whether or not the Offer is consummated, while it is pending we will continue to incur costs, fees, expenses and charges related to the Offer, which may materially and adversely affect our financial condition and results of operations.
In addition, while the Purchase Agreement generally requires the Company to operate its business in the ordinary course of business consistent with past practice pending consummation of the Offer, it also restricts us from taking certain actions with respect to our business and financial affairs without Thermo Fisher’s consent. Such restrictions will be in place until either the Offer is consummated or the Purchase Agreement is terminated. For these and other reasons, the pendency of the Offer could adversely affect our business and results of operations.
In the event that the Offer is not consummated, the trading price of our common stock and our future business and results of operations may be negatively affected.
The conditions to the consummation of the Offer may not be satisfied as noted above. If the Offer is not consummated, we would remain liable for significant transaction costs, and the focus of our management would have been diverted from seeking other potential strategic opportunities, in each case without realizing any benefits of the Offer. For these and other reasons, not consummating the Offer could adversely affect our business and results of operations. Furthermore, if we do not consummate the Offer, the price of our common stock may decline significantly from the current market price, which we believe reflects a market assumption that the Offer will be consummated. Certain costs associated with the Offer have already been incurred or may be payable even if the Offer is not consummated. Further, a failed Offer may result in negative publicity and a negative impression of us in the investment community. Finally, any disruptions to our business resulting from the announcement and pendency of the Offer, including any adverse changes in our relationships with our customers, vendors and employees or recruiting and retention efforts, could continue or accelerate in the event of a failed acquisition.
Risks Related to our Business and Industry
If we do not successfully manage the development, launch and scaling of new products, including our Explore product line, our Olink Signature platform, our Olink Flex and Olink Insight initiatives, our financial results could be adversely affected.
In June 2020, we introduced our Explore product line to the market. We face risks associated with launching new products, such as new Explore products, and platforms, such as our Olink Signature Q100, which we started delivering to customers in the fall of 2021, both leading up to such a launch and also for some time following the launch. During the fourth quarter of 2022 we launched our Olink Insight and Olink Flex initiatives to further enable our Target Kit strategy as well as our data sharing and collaboration initiatives. If we encounter development, manufacturing, performance or scaling challenges our anticipated growth may be hindered. The expenses or losses associated with unsuccessful product development, launch activities, or scaling opportunities, or lack of market acceptance of our new products could adversely affect our business or financial condition.
We are substantially dependent on the success of scaling our distributed kits model through Explore, Target, Flex and Olink Signature. If we are unable to successfully roll out and scale this business model, our business will be materially harmed.
/7
To date, we have invested significant efforts and financial resources in the development of our Explore product line offering to enable a scalable distributed kits model, which we began delivering to early access customers in 2020 followed by a full commercial launch in March 2021, and the Olink Signature platform, which we started shipping to customers in the fall of 2021. Our near-term prospects, including our continued ability to finance our operations and generate revenue, will depend substantially on the successful performance of our Explore and Target kits sales, as well as adoption of our Olink Signature platform. During 2022 our Target kit offering was expanded through the introduction of Olink Flex, which we started shipping to customers late 2022, and during 2023 our Explore Kit was expanded through the introduction of Olink Explore HT, which we started shipping to customers mid 2023. The commercial success of our distributed kits will depend on a number of factors, including:
•our ability to gain traction for our external installations, scaling our footprint to enable the transition to a more distinct distributed kits business model;
•the consistent supply of the necessary equipment and consumables required for the Proximity Extension Assay, or PEA, workflows to our customers by third-party vendors;
•the ability of our customers to secure any necessary internal approvals, and in some cases financing, to adopt the technology;
•the accessibility of Illumina’s NGS technology, which is the underlying readout platform for Explore;
•the availability, perceived advantages, relative cost, and relative performance of alternative and competing products;
•the effectiveness of our own or any future strategic collaborators’ marketing, sales and distribution strategy and operations;
•our ability to obtain, maintain, protect, and enforce our intellectual property rights in and to our Explore product line and our Olink Signature platform;
•our ability to avoid and defend against third-party patent interference or patent infringement claims or other intellectual property-related claims; and
•our ability to raise sufficient capital resources to fund the continued commercialization and roll out of the entire Olink product platform.
Many of these factors are beyond our control. If we are not successful with respect to one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize our distributed kits model, which would materially harm our business. If we are not successful in commercializing our Explore kits or Olink Signature platform, our business will be materially harmed.
If we do not successfully develop and introduce new assays for our technology, we may not generate new sources of revenue and may not be able to successfully implement our growth strategy.
Our business strategy includes the development of new assays for our library of protein biomarker targets. New assays require significant research and development and a commitment of significant resources prior to their commercialization. Our technology is complex, and we cannot be sure that any assays we intend to develop will be developed successfully, be proven to function as intended, offer improvements over currently available tests, meet applicable standards, be produced in commercial quantities at acceptable costs or be successfully marketed. We cannot assure you that any assays we develop will be manufactured or produced economically, successfully commercialized or widely accepted in the marketplace or be more effective than other commercially available alternatives. Moreover, development of particular assays may require licenses or access to third-party intellectual property which may not be available on commercially reasonable terms, or at all. If we do not successfully develop new high-multiplex assays for our protein biomarker targets, we could lose revenue opportunities with existing or future customers.
Our long-term results depend upon our ability to improve existing products and introduce and market new products successfully.
Our business is dependent on the continued improvement of our existing products and our development of new products utilizing our existing or potential future technology. As we introduce new products or refine, improve, or upgrade versions of existing products, we cannot predict the level of market acceptance or the amount of market share these products will achieve, if any. We cannot assure you that we will not experience material delays in the introduction of new products or
/8
that evolving supply chains will not be materially delayed or disrupted in the future. In addition, introducing new products could result in a decrease in revenues from our existing products. Consistent with our strategy of offering new products and product refinements, we expect to continue to use a substantial amount of capital for product development and refinement. We may need more capital for product development and refinement than is available on terms favorable to us, if at all, which could adversely affect our business, financial condition, or results of operations.
We generally sell our products in industries that are characterized by rapid technological changes, frequent new product introductions and changing industry standards. If we do not develop new products and product enhancements based on technological innovation on a timely basis, our products may become obsolete over time and our revenues, cash flow, profitability and competitive position will suffer. Our success will depend on several factors, including our ability to:
•correctly identify customer needs and preferences and predict future needs and preferences;
•allocate our research and development funding to products with higher growth prospects;
•anticipate and respond to our competitors’ development of new products and technological innovations;
•innovate and develop new technologies and applications, and acquire or obtain rights to third-party technologies that may have valuable applications in the markets we serve;
•successfully commercialize new technologies in a timely manner, price them competitively and manufacture and deliver sufficient volumes of new products of appropriate quality on time;
•maintain our existing collaborative relationships with key opinion leaders (KOLs) in the life sciences scientific community;
•convince customers to adopt new technologies; and
•develop functioning global supply chains with multiple third-parties to bring products to market.
In addition, if we fail to accurately predict future customer needs and preferences or fail to produce viable technologies, we may invest heavily in research and development of products that do not lead to significant revenue. Even if we successfully innovate and develop new products and product enhancements, we may incur substantial costs in doing so, and our profitability may suffer.
Our ability to develop new products based on innovation can affect our competitive position and often requires the investment of significant resources. Difficulties or delays in research, development or production of new products and services or failure to gain market acceptance of new products and technologies may reduce future revenues and adversely affect our competitive position.
We have estimated the sizes of the markets for our current and future products and services, and these markets may be smaller than we estimate.
The market for proteomics technologies and products is new and evolving, making it difficult to predict with any accuracy the size of the markets for our current and future products. Our estimates of the total addressable market for our current products and services and those under development are based on a number of internal and third-party estimates, including, without limitation, the research community’s unmet need for methods to better facilitate prediction of drug response and disease risk and outcomes, whether novel proteomics are successfully integrated into the genomics markets from full discovery to clinical decision making, the applicability of our technology in vitro diagnostics and laboratory developed tests, and the assumed prices at which we can sell our current and future products and services for markets that have not been established. While we believe our assumptions and the data underlying our estimates are reasonable, these assumptions and estimates may not be correct and the conditions supporting our assumptions or estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. As a result, our estimates of the total addressable market for our current or future products and services may prove to be incorrect.
/9
The future growth of the market for our current and future products depends on many factors beyond our control, including recognition and acceptance of our products by the scientific community and the growth, prevalence and costs of competing products and solutions. Such recognition and acceptance may not occur in the near term, or at all. If the markets for our current and future products are smaller than estimated or do not develop as we expect, or if the price at which we can sell future products and services or the total addressable market for our products or services is smaller than we have estimated, our growth may be limited and our business, financial condition and results of operations could be adversely affected.
The life science tools markets are highly competitive. If we fail to effectively compete, our business, financial condition and operating results will suffer.
We face significant competition in the life science tools markets. We currently compete with both established and early- stage life science tools companies that design, manufacture and market assay products and services and libraries of protein biomarker targets. We believe our principal competitors in the life science tools markets as a whole are Quanterix Corporation, Meso Scale Diagnostics LLC, Luminex Corporation and SomaLogic Inc. as well as more established technologies such as ELISA or mass spectrometry provided by a number of established vendors. In addition, there are a number of new market entrants, such as Alamar, Seer Inc., Encodia, Nautilus Biotechnology, Spear Bio Inc. and Quantum-Si Incorporated, in the process of developing novel technologies for the life sciences market, including those that may compete with our PEA technology and existing product lines. Depending on market segment and customer use-case the relevant competitors may vary.
Some of our current competitors are large, well-capitalized, publicly-traded companies, or are divisions of large, well-capitalized, publicly- traded companies, and may enjoy a number of competitive advantages over us, including:
•greater name and brand recognition, financial and human resources;
•larger sales forces and more established distributor networks;
•substantial intellectual property portfolios;
•larger libraries of protein biomarkers; and
•better established, larger scale, and lower cost manufacturing capabilities.
We believe that the principal competitive factors in all of our target markets include:
•market adoption;
•scientific proof;
•cost of capital equipment;
•cost of consumables and supplies;
•reputation among customers and KOLs;
•innovation in product offerings;
•flexibility and ease-of-use;
•accuracy and reproducibility of results; and
•compatibility with existing laboratory processes, tools, and methods.
/10
We cannot assure investors that our products will compete favorably or that we will be successful in the face of increasing competition from new products and technologies introduced by our existing competitors or new companies entering our markets. In addition, we cannot assure investors that our competitors do not have or will not develop products or technologies that currently or in the future will enable them to produce competitive products with greater capabilities or at lower costs than ours. Although we are pursuing several strategies to mitigate this trend, there can be no assurance we will be successful in doing so. Any failure to compete effectively could materially and adversely affect our business, financial condition, and operating results.
Our business depends on levels of research and development spending by academic and governmental research institutions and biopharmaceutical companies, a reduction in which could limit demand for our products and adversely affect our business and operating results.
In the near term, we expect that a vast majority of our revenue will be derived from sales of the following product lines: Explore, Target (Including Signature), and Focus, including our Signature platform, to academic and clinical institutions and biopharmaceutical and biotechnology companies worldwide for research and development applications. The demand for our products will depend in part upon the research and development budgets of these customers, which are impacted by factors beyond our control, such as:
•changes in government programs (such as the National Institutes of Health) that provide funding to research institutions and companies;
•macroeconomic conditions (including inflation), the political climate, and any lingering impacts from the COVID-19 pandemic;
•changes in the regulatory environment;
•differences in budgetary cycles;
•competitor product offerings or pricing;
•market-driven pressures to consolidate operations and reduce costs; and
•market acceptance of relatively new products.
In addition, academic, governmental, and other research institutions that fund research and development activities may be subject to stringent budgetary constraints that could results in spending reductions, reduced allocations, or budget cutbacks, which could jeopardize the ability of these customers to purchase our products. Our operating results may fluctuate substantially due to reductions and delays in research and development expenditures by these customers. We cannot assure investors that any changes to our customers’ spending patterns are temporary or whether any new spending patterns will be sustained. Any decrease in our customers’ budgets or expenditures, or in the size, scope, or frequency of capital or operating expenditures, could materially and adversely affect our business, operating results, and financial condition.
If we cannot provide quality technical and applications support, we could lose customers and our business and prospects will suffer.
The placement of our products and third-party instruments used with our products at new customer sites, the introduction of our technology into our customers’ existing laboratory workflows and ongoing customer support can be complex. Accordingly, we need highly trained technical support personnel. Hiring technical support personnel is very competitive in our industry due to the limited number of people available with the necessary scientific and technical backgrounds and ability to understand our technology at a technical level. To effectively support potential new customers and the expanding needs of current customers, we will need to substantially expand our technical support staff and develop our support
/11
infrastructure and processes. If we are unable to attract, train or retain the number of highly qualified technical services personnel that our business needs, our business, and prospects will suffer.
We may experience manufacturing problems or delays that could limit our growth or adversely affect our operating results.
Our products are manufactured at our facilities located in Uppsala, Sweden and Umeå, Sweden using complex processes, sophisticated equipment and strict adherence to specifications and quality systems procedures. Any unforeseen manufacturing problems, such as contamination of our facilities, equipment malfunction, quality issues with components and materials sourced from third-party suppliers (such as our OEM partner for our Signature platform), failure to strictly follow procedures or meet specifications, or limitations on access to our facilities, could result in delays or shortfalls in production or require us to voluntarily recall our products. Identifying and resolving the cause of any such manufacturing or supplier issues could require substantial time and resources. If we are unable to keep up with demand for our products by successfully manufacturing and shipping our products in a timely manner, our revenue could be impaired, market acceptance for our products could be adversely affected and our customers might instead purchase our competitors’ products or cancel outstanding purchase orders.
In addition, the introduction of new products may require the development of new manufacturing sites and processes or procedures as well as new suppliers. While all of our assays are currently produced using the same basic processes, significant variations may be required to meet new product specifications.
Developing new processes and negotiating supply agreements can be very time consuming, and any unexpected difficulty in doing so could delay the introduction of a product.
Undetected errors or defects in our products, services and software could harm our reputation and decrease market acceptance of our products, services, and software.
Our products and services, as well as the software that accompanies them, are novel and complex and may contain undetected errors or defects when first introduced or as new versions are released. We cannot assure you that material performance problems, defects, or errors will not arise, and as we commercialize our Olink Signature platform with new software and launch more applications and content on Olink Insight, these risks may increase. We provide warranties that our products will meet performance specifications and will be free from defects. The costs incurred in correcting any defects or errors may be substantial and could adversely affect our operating margins.
In manufacturing our products, we depend upon third parties for the supply of various components, many of which require a significant degree of technical expertise to produce. If our suppliers fail to produce our components to specification or provide defective products to us and our quality control tests and procedures fail to detect such errors or defects, or we or our suppliers use defective materials in the manufacturing process, the reliability and performance of our products will be compromised.
Disruptions or other performance problems with our products, services or software may adversely impact our customers’ research or business, harm our reputation and result in reduced revenue or increased costs associated with product repairs or replacements. If that occurs, we may also incur significant costs, the attention of our key personnel could be diverted or other significant customer relations problems may arise.
We may be subject to claims related to errors or defects in our products, services, or software.
Errors or defects in our products, services or software may give rise to claims against us that exceed any revenue or profit we receive from the affected products, services, or software. Our limited representations for services cover nonconformance with generally accepted and applicable standards of service, and our limited product warranties cover manufacturing defects for use in accordance with applicable specifications and instructions.
/12
Any lingering impacts of the COVID-19 pandemic may create significant uncertainty for our business, financial condition, and results of operations notwithstanding the easing of government-mandated restrictions and could continue to adversely impact our business.
The extent of any lingering impacts of the COVID-19 pandemic on our business and financial results will continue to depend on numerous evolving factors that we are not able to accurately predict and which will vary by market, including new surges in the spread of COVID-19, the pandemic's impact on global economic conditions, governmental actions that may be taken in the future, in response to resurgences of the pandemic, and changes in customer behaviors during the pandemic that may continue on beyond the end of the pandemic. Our global operations expose us to risks associated with the COVID-19 pandemic, which may result in challenging operating environments. COVID-19 has spread across the globe to almost all countries and territories in which our products are developed, made, manufactured, distributed or sold. Authorities in many of these countries and territories have implemented or may resume numerous measures to stall the spread and reduce the impact of COVID-19, including travel bans and restrictions, quarantines, curfews, shelter in place and safer-at-home orders, business shutdowns and closures, and have also implemented multi-step polices with the goal of re-opening these markets. These measures have impacted and may continue to impact us, our employees, customers, manufacturers, distributors, partners, suppliers and other third parties with whom we do business. Lingering impacts from the pandemic may adversely affect elements of our business.
We primarily observed disruptions in the customer end of the supply chain, with our customers’ labs operating at reduced capacity for extended parts of 2020 and 2021, and during 2021 and 2022 we primarily observed continued disruptions in our supply chain related to standard lab consumables. COVID-19 adversely impacted our growth rate for 2020 and 2021, in particular as customers have had issues accessing their labs. We have not seen any material cancellations in our pipeline; however, there have been delays with projects being pushed into the future. We are continuing to closely monitor how the pandemic and related response measures are affecting our business. Our production and manufacturing facilities are located in Uppsala, Sweden; Umeå, Sweden, and Waltham, Massachusetts, and we have noted a continued increase in delivery times for certain components throughout our supply chain. There is a risk that we could experience continued disruption on the supply side beyond the end of the pandemic. The recovery of revenue we have seen compared with previous levels reflects the underlying factors affecting demand, including the easing of lockdown restrictions and the partial or full reopening of academic and biopharmaceutical research laboratories around the world. As of December 31, 2023, we concluded that there was no evidence of material changes in the recovery risk of business assets, including deferred tax assets and trade receivables. In light of the total duration of the risk factor and that COVID-19 is still widespread and if a new variant flares up, and the government restrictions may reappear, we continue to follow this closely.
The countries and territories in which our products are developed, made, manufactured, distributed or sold vary in their stages of restrictions to address the COVID-19 pandemic. Certain jurisdictions re-opened only to return to restrictions in the face of increases in COVID-19 cases and new variants. There is still considerable uncertainty regarding how the effects of the pandemic, including any future health and safety measures that was implemented in response to the pandemic, may continue to affect our business, including whether they will result in further changes in demand for our products further increases in operating costs (whether as a result of changes to our supply chain or increases in employee costs, operating costs or otherwise); further impact our ability to perform research and development, manufacturing, and shipping of our products how they will further impact our supply chain and whether they will result in further reduced availability of air or other commercial transport, port closures or border restrictions, each or all of which can impact our ability to make, manufacture, distribute and sell our products. In addition, measures that impact our ability to access our facilities may continue to impact the availability of our employees, some of whom are not able to perform their job functions remotely. If a significant percentage of our or our business partners’ workforce is unable to work (including because of illness, facility closures, quarantine, curfews, shelter in place orders, travel restrictions, social distancing requirements or other governmental restrictions or voluntarily adopted practices), our operations will be negatively impacted. Any sustained interruption in our or our business partners’ operations, research and development, distribution network or supply chain or any significant continuous shortage of raw materials or other supplies as a result of these measures, restrictions or disruptions, including as a result of increased demand for certain products, can impair our ability to develop, make, manufacture, distribute or sell our products.
Compliance with governmental measures imposed in response to COVID-19 has caused and may continue to cause us to incur additional costs, and any failure to comply with such measures could subject us to restrictions on our business operations, fines and other penalties, which preferable can affect our business negatively. In addition, the increase in certain of our employees working remotely has increased certain risks to our business, including increased demand on our information technology resources and systems, increased phishing and other malicious activity as cybercriminals seek to exploit the uncertainty surrounding the pandemic and its lingering impacts and an increase in the number of points of potential exposure, such as laptops and mobile devices, to be secured, and any failure to effectively manage these risks, including to timely identify and appropriately respond to any security incidents, could continue to adversely affect our business.
/13
Even as governmental restrictions have been lifted and economies re-opened, the ongoing economic impacts and health concerns associated with the pandemic may continue to affect customer behavior. In addition, changes in customer purchasing patterns may increase demand for our products in one quarter, resulting in decreased customer demand for our products in subsequent quarters. Additionally the pandemic created volatility in the global capital and credit markets which also hereafter could impair our ability to access these markets on terms commercially acceptable to us, or at all, and execute our growth strategies. While we have developed and implemented and continue to develop and implement health and safety protocols, business continuity plans and crisis management protocols in an effort to try to mitigate that any lingering negative impacts of the pandemic on our employees and our business, there can be no assurance that we will be successful in our efforts or that such efforts may not have detrimental unintended consequences, and as a result, our business, financial condition and results of operations and the price of our common shares and ADSs may continue to be adversely affected.
Our products could become subject to government regulation, and the regulatory approval and maintenance process for such products may be expensive, time-consuming and uncertain in both timing and outcome.
Our products are currently labeled and promoted, and are, and in the near-future will be, sold primarily to academic and research institutions and biopharmaceutical companies as research use only (RUO) products, and are not currently designed, or intended to be used, for clinical diagnostic tests. However, as we continue to expand our product lines and the applications and uses of our existing products into new fields, certain of our current or future products could become subject to regulation by the United States Food and Drug Administration (FDA), European Medicines Agency (EMA), or comparable international agencies, including requirements for regulatory clearance, authorization or approval of such products before they can be marketed. Also, even if our products are labeled, promoted and intended as RUO, the FDA, EMA or comparable international agencies could disagree with our conclusion that our products are intended for research use only or deem our sales, marketing and promotional efforts as being inconsistent with RUO products. For example, our customers may independently elect to use our RUO labeled products in their own LDTs for clinical diagnostic use, which could subject our products to government regulation, even if clinical uses of our RUO products by our customers were done without our consent. Such regulatory approvals, authorizations or clearances may be expensive, time-consuming and uncertain, and our failure to obtain or comply with such approvals, authorizations and clearances could have an adverse effect on our business, financial condition and operating results. In addition, changes to the current regulatory framework, including the imposition of additional or new regulations, including regulation of our products, could arise at any time during the development or marketing of our products, which may negatively affect our ability to obtain or maintain FDA, EMA or comparable regulatory approval of our products, if required. Also, obtaining and maintaining marketing approval of our current and future products in one jurisdiction does not mean that we will be successful in obtaining marketing approval of our current and future product candidates in other jurisdictions. Further, if we expand into new product lines or services, we may become subject to additional U.S. healthcare regulations such as federal and state fraud and abuse, transparency and data privacy and security laws and state clinical laboratory requirements, among others.
Diagnostic products are regulated as medical devices by the FDA, EMA and comparable international agencies and may require clearance following the 510(k)-pre-market notification process, authorization following a request for de novo classification or pre-market approval from the FDA, in each case prior to marketing. In Europe, we are required to comply with the Medical Device Regulation 2017/745 and In Vitro Diagnostic Regulation 2017/746, which became effective May 26, 2017, with application dates of May 26, 2021 (postponed from 2020) and May 26, 2022, respectively. Obtaining the requisite regulatory approvals can be expensive and may involve considerable delay. None of our products are currently regulated as in vitro diagnostic devices for clinical diagnosis. However, if our products labeled as RUO are used, or could be used, for the diagnosis of disease, the regulatory requirements related to marketing, selling and supporting such products could change or be uncertain, even if such use by our customers is without our consent. Moreover, if the FDA believed we inappropriately labeled our products as RUO, it could allege that we had misbranded or adulterated our products.
If the FDA, EMA or other regulatory authorities assert that any of our products are subject to regulatory clearance, authorization or approval, our business, financial condition or results of operations could be adversely affected.
The raw materials for and components of our products could become subject to stricter regulation.
Antibodies are a key component of our products. The Scientific Advisory Committee (ESAC) of the European Union Reference Laboratory for alternatives to animal testing (EURL ECVAM) published a recommendation in May 2020 on non- animal derived antibodies which, in summary, stated that animals should no longer be used for the development and production of antibodies for research, regulatory, diagnostic and therapeutic applications and that countries in the European Union should no longer authorize the development and production of antibodies through animal immunization, where robust, legitimate scientific justification is lacking. The recommendation is based on the principle from European Union Directive 2010/63 on the protection of animals used for scientific purposes, that European Union Member States should ensure that, wherever possible, a scientifically satisfactory method or testing strategy not entailing the use of live animals should be used over any procedure that may be harmful to animals. The ESAC recommendation suggests that non-animal derived antibodies
/14
are equivalent to animal-derived antibodies for the vast majority of applications and encourages manufacturers and suppliers to replace animal-derived antibodies available in their catalogues with non-animal-derived affinity reagents. While the ESAC recommendation is not legally-binding, and its principles are yet to be enacted in legislation, it does suggest a policy move away from the use of animal immunization for developing and producing antibodies in the European Union and, in particular, that European Union Member States may need to adapt their national regulations on antibody development and production to ensure compliance with Directive 2010/63. This may result in stricter regulation in the future which could have an adverse impact on our operations and antibody suppliers. Regulation (EU)2019/1010 introduced a new level of transparency to help progress towards eventually replacing animal use in science. The amendments have been incorporated in the consolidated text of the Directive 2010/63.
We face risks related to handling of hazardous materials and other regulations governing environmental safety.
Our operations are subject to complex and stringent environmental, health, safety and other governmental laws and regulations that both public officials and private individuals may seek to enforce. Our activities that are subject to these regulations include, among other things, our use of hazardous materials in manufacturing and in our products, and the generation, transportation and storage of waste. We could discover that we, an acquired business or our suppliers are not in material compliance with these regulations. Existing laws and regulations may also be revised or reinterpreted, or new laws and regulations may become applicable to us, whether retroactively or prospectively, that may have a negative effect on our business and results of operations. It is also impossible to eliminate completely the risk of accidental environmental contamination or injury to individuals. In such an event, we could be liable for any damages which could adversely affect our business.
Acquisitions or joint ventures could disrupt our business, cause dilution to our shareholders and/or our holders of ADSs and otherwise harm our business.
We may acquire other businesses, products or technologies as well as pursue strategic alliances, joint ventures, technology licenses or investments in complementary businesses. For example, in early 2020, we acquired Agrisera AB, a Swedish company specializing in antibody production, in order to enable the growth of our protein biomarker library and increase control over our supply chain. Any future transactions could be material to our financial condition and operating results and expose us to many risks, including:
•disruption in our relationships with customers, distributors or suppliers as a result of such a transaction;
•unanticipated liabilities related to acquired companies;
•difficulties integrating acquired personnel, technologies and operations into our existing business;
•diversion of management time and focus from operating our business;
•increases in our expenses and reductions in our cash available for operations and other uses; and
•possible write-offs or impairment charges relating to acquired businesses.
Foreign acquisitions involve unique risks in addition to those mentioned above, including those related to integration of operations across different cultures and languages, currency risks and the particular economic, political and regulatory risks associated with specific countries.
Also, the anticipated benefit of any strategic transaction may not materialize. Future acquisitions could result in potentially dilutive issuances of our equity securities, the incurrence of debt, contingent liabilities or amortization expenses or write-offs of goodwill, any of which could harm our financial condition. We cannot predict the number, timing or size of future joint ventures or acquisitions, or the effect that any such transactions might have on our operating results.
Unfavorable global economic or political conditions could adversely affect our business, financial condition or results of operations.
General conditions in the global economy and in the global financial markets could adversely affect our results of operations, including the potential effects from COVID-19 as discussed above as well as the effects of increased inflation and cost of capital, and the overall demand for our products and services may be particularly vulnerable to unfavorable economic conditions. A global financial crisis or a global or regional political disruption could cause extreme volatility in the capital and credit markets. A severe or prolonged economic downturn or political disruption could result in a variety of risks to our business, including weakened demand for our products and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy or political disruption could also strain our manufacturers or suppliers, possibly
/15
resulting in supply disruption, or cause our customers to delay making payments for our products and services. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the political or economic climate and financial market conditions could adversely impact our business.
Enhanced trade tariffs, import restrictions, export restrictions, United States regulations, Chinese regulations or other trade barriers may materially harm our business.
We are continuing to expand our international operations as part of our growth strategy and have experienced an increasing concentration of sales in certain regions, especially in the Asia-Pacific region. These regions, including China, could impose tariffs on imports from various regions, including from regions where we operate our business, and these tariffs could raise our costs. Furthermore, tariffs, trade restrictions, or trade barriers that have been, and may in the future be, placed on products such as ours by foreign governments, especially China, have raised, and could further raise, amounts paid for some or all of our products, which may result in the loss of customers and our business, and our financial condition and results of operations may be harmed. Further tariffs may be imposed that could cover imports of components and materials used in our products, or our business may be adversely impacted by retaliatory trade measures taken by China or other countries, including restricted access to components or materials used in our products or increased amounts that must be paid for our products, which could materially harm our business, financial condition and results of operations. Further, the continued threats of tariffs, trade restrictions and trade barriers could have a generally disruptive impact on the global economy and therefore, negatively impact our sales. Given the relatively fluid regulatory environment in China and uncertainty how foreign governments will act with respect to tariffs, international trade agreements, restrictions and policies, there could be additional tax or other regulatory changes in the future. Any such changes could directly and adversely impact our financial results and results of operations.
Additionally, in November 2018, the United States Commerce Department’s Bureau of Industry and Security (BIS) released an advance notice of proposed rulemaking to control the export of emerging technologies. This notice included “biotechnology, including nanobiology; synthetic biology; genomic and genetic engineering; or neurotech” as possible areas of increased export controls. In April 2020, BIS expanded its controls on the export, re-export, and transfer of certain items for military end-use or to military end-users in China and certain other countries. Therefore, it is possible that our ability to export or share our technologies developed in the United States may be restricted in the future.
Risks Related to Our Financial Position and Need for Additional Capital
We expect to make significant investments in our continued research and development of new products and services and software, which may not be successful.
We currently have a library more than 5,400 protein biomarker targets, of which 1,100 are incorporated in the Target product line and all 5,400 are incorporated in Explore as of the second quarter 2023. We plan to grow our library as far as determined from a commercial and scientific perspective over time. We continue to plan to make our Explore line widely available as distributed kit products and to continue the roll out of our own qPCR readout platform, Olink Signature Q100. In addition, we plan to utilize our cloud platform, Olink Insight, and work together with KOLs and our customers to make proteomics big data easy, accessible and actionable, which in turn requires open access, transparent and high-quality protein biomarker data. We also plan to invest in our sales and marketing infrastructure to grow our customer base and sell more products and services to existing customers. We expect to incur significant expenses to advance these development efforts, but they may not be successful. Even if we are ultimately successful in these efforts, our gross margins may suffer as we invest in advance of potential revenue growth.
Developing new products, services and software is a speculative and risky endeavor. Products, services or software that initially show promise may fail to achieve the desired results or may not achieve acceptable levels of analytical accuracy or clinical utility. We may need to alter our products in development and repeat studies before we identify a potentially successful product or service. Product development is expensive, may take years to complete and can have uncertain outcomes. Failure can occur at any stage of the development. If, after development, a product appears successful, we or our collaborators may, depending on the nature of the product, need to obtain FDA, EMA and other regulatory clearances, authorizations or approvals before we can market the product. The FDA’s and EMA’s clearance, authorization or approval pathways are likely to involve significant time, as well as additional research, development and clinical study expenditures. The FDA, EMA or other applicable regulatory authority may not clear, authorize or approve any future product we develop. Even if we develop a product that receives regulatory clearance, authorization or approval, we or our collaborators would need to commit substantial resources to commercialize, sell and market the product before it could be profitable, and the product or service may never be commercially successful. Additionally, development of any product or service may be disrupted or made less viable by the development of competing products or services.
New potential products, services and software may fail at any stage of development or commercialization and if we determine that any of our current or future products, services or software is unlikely to succeed, we may abandon them
/16
without any return on our investment. If we are unsuccessful in developing additional products, services or software, our potential for growth may be impaired.
Our future capital needs are uncertain and we may need to raise additional funds in the future.
We believe that our existing cash at bank and in hand as of December 31, 2023, together with our cash generated from commercial sales, will enable us to fund our operating expenses and capital expenditure requirements for at least the next 12 months. However, we may need to raise substantial additional capital to:
•expand our sales and marketing efforts to further commercialize our products;
•strategically acquire companies or technologies that may be complementary to our business;
•expand our research and development efforts to improve our existing products and develop and launch new products, particularly if any of our products are deemed by the FDA, EMA or other applicable regulatory authority to be medical devices or otherwise subject to additional regulation by the FDA, EMA or other applicable regulatory authority;
•seek premarket approval, de novo classification or 510(k) clearance from the FDA and comply with the new Medical Device Regulation 2017/745 and In Vitro Diagnostic Regulation 2017/746 in Europe for our existing products or new products if or when we decide to market products for use in the prevention, diagnosis or treatment of a disease or other condition (see “- Our products could become subject to government regulation and the regulatory approval and maintenance process for such products may be expensive, time-consuming and uncertain in both timing and outcome” for further information about the FDA, EMA and other regulatory approvals that we may be required to seek and obtain in that circumstance);
•hire additional personnel;
•enter into collaboration arrangements, if any, or in-license other products and technologies;
•add operational, financial and management information systems; and
•pay for increased costs as a result of operating as a public company.
Our future funding requirements will depend on many factors, including:
•market acceptance of new products, including our recently launched Explore product line and our future products;
•the cost and timing of establishing additional sales, marketing and distribution capabilities;
•the cost of our research and development activities;
•our ability to enter into collaborations in the future, and the success of any such collaborations;
•the cost and timing of potential regulatory clearances, authorizations or approvals that may be required in the future for our products; and
•the effect of competing technological and market developments.
We cannot assure you that we will be able to obtain additional financing for investment for growth on acceptable terms, or at all. Our ability to raise additional funds will depend on financial, economic and market conditions and other factors, over which we may have no or limited control. Market volatility resulting from increased inflation and interest rates or other factors could also adversely impact our ability to access capital as necessary. If we raise additional funds by issuing equity or equity-linked securities, our shareholders and future holders of the ADSs may experience dilution. Future debt financing, if available, may involve covenants restricting our operations or our ability to incur additional debt. Any debt or equity financing may contain terms that are not favorable to us, our shareholders or future holders of the ADSs. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish some rights to our technologies or our products or grant licenses on terms that are not favorable to us. If we do not have, or are not able to obtain, sufficient funds, we may have to delay development or commercialization of new products. We also may have to reduce marketing, customer support or other resources devoted to our products or cease operations. Any of these factors could have a material adverse effect on our financial condition, operating results and business.
/17
Adverse developments affecting the financial services industry, such as actual events or concerns involving liquidity, defaults, or non-performance by financial institutions or transactional counterparties, could adversely affect the Company’s current and projected business operations and its financial condition and results of operations.
Actual events involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions, transactional counterparties or other companies in the financial services industry or the financial services industry generally, or concerns or rumors about any events of these kinds or other similar risks, have in the past and may in the future lead to market-wide liquidity problems. For example, on March 10, 2023, Silicon Valley Bank (“SVB”) was closed by the California Department of Financial Protection and Innovation, which appointed the Federal Deposit Insurance Corporation (“FDIC”) as receiver. Similarly, on March 12, 2023, Signature Bank and Silvergate Capital Corp. were each swept into receivership. Although we are not a borrower or party to any such instruments with any financial institution currently in receivership, if any financial institution with whom we bank or borrow money were to be placed into receivership, we may be unable to access such funds. In addition, if any of our customers, suppliers or other parties with whom we conduct business are unable to access funds from a financial institution placed into receivership, such parties’ ability to pay or perform their obligations or to enter into new commercial arrangements requiring additional payments to us could be adversely affected.
The results of events or concerns that involve one or more of these factors could include a variety of material and adverse impacts on our current and projected business operations and our financial condition and results of operations. These could include, but may not be limited to, the following:
1.Delayed access to deposits or other financial assets or the uninsured loss of deposits or other financial assets;
2.Delayed or lost access to, or reductions in borrowings available under revolving existing credit facilities or other working capital sources and/or delays, inability or reductions in the company’s ability to refund, roll over or extend the maturity of, or enter into new credit facilities or other working capital resources;
3.Potential or actual breach of contractual obligations that require the Company to maintain letters of credit or other credit support arrangements;
4.Potential or actual breach of financial covenants in our credit agreements or credit arrangements;
5.Potential or actual cross-defaults in other credit agreements, credit arrangements or operating or financing agreements; or
6.Termination of cash management arrangements and/or delays in accessing or actual loss of funds subject to cash management arrangements.
7.Our inability to finance future business or growth opportunities.
In addition, investor concerns regarding the U.S. or international financial systems could result in less favorable commercial financing terms, including higher interest rates or costs and tighter financial and operating covenants, or systemic limitations on access to credit and liquidity sources, thereby making it more difficult for us to acquire financing on acceptable terms or at all. Any decline in available funding or access to our cash and liquidity resources could, among other risks, adversely impact our ability to meet our operating expenses, financial obligations or fulfill our other obligations, result in breaches of our financial and/or contractual obligations or result in violations of federal or state wage and hour laws. Any of these impacts, or any other impacts resulting from the factors described above or other related or similar factors not described above, could have material adverse impacts on our liquidity and our current and/or projected business operations and financial condition and results of operations.
In addition, any further deterioration in the macroeconomic economy or financial services industry could lead to losses or defaults by our customers or suppliers, which in turn, could have a material adverse effect on our current and/or projected business operations and results of operations and financial condition. For example, a customer may fail to make payments when due, default under their agreements with us, become insolvent or declare bankruptcy, or a supplier may determine that it will no longer deal with us as a customer. In addition, a customer or supplier could be adversely affected by any of the liquidity or other risks that are described above as factors that could result in material adverse impacts on the Company, including but not limited to delayed access or loss of access to uninsured deposits or loss of the ability to draw on existing credit facilities involving a troubled or failed financial institution. Any customer or supplier bankruptcy or insolvency, or the failure of any customer to make payments when due, or any breach or default by a customer or supplier, or the loss of any significant supplier relationships, could result in material losses to the Company and may have a material adverse impact on our business.
We have incurred net losses, from time to time since we were formed, and we may incur losses in the future.
/18
We recorded revenue of $169.6 million and $139.8 million; and recognized net losses of $31.6 million and $12.9 million during the year ended December 31, 2023, and December 31, 2022, respectively. We might continue to incur losses in the future as we plan to invest significant additional funds toward expansion of our commercial organization and the development of our technology. In addition, as a public company, we will incur significant legal, accounting, and other expenses that we did not incur as a private company. These increased expenses will make it harder for us to sustain future profitability. We may incur losses in the future for a number of reasons, many of which are beyond our control, including the other risks described in this “Risk Factors” section, the market acceptance of our new products, future product development and our market penetration and margins. Our failure to become profitable would depress the value of our common shares and ADSs and could impair our ability to raise capital, expand our business, maintain our research and development efforts or continue our operations. A decline in the value of our common shares or ADSs could also cause you to lose all or part of your investment.
/19
We have a limited operating history, which may make it difficult to evaluate the prospects for our future viability and predict our future performance.
Our operations to date have been limited to developing and commercializing our technology and products. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. Predictions about our future success or viability are highly uncertain and may not be as accurate as they could be if we had a longer operating history. In addition, as a business with a limited operating history, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown obstacles. We have encountered in the past, and will encounter in the future, risks and uncertainties frequently experienced by growing companies with limited operating histories in emerging and rapidly changing industries. If our assumptions regarding these risks and uncertainties, which we use to plan and operate our business, are incorrect or change, or if we do not address these risks successfully, our results of operations could differ materially from our expectations, and our business, financial condition and results of operations could be adversely affected.
Our operating results have in the past fluctuated significantly and may continue to fluctuate significantly in the future, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or any guidance we may provide.
Our quarterly and annual operating results have fluctuated significantly, which makes it difficult for us to predict our future operating results. These fluctuations have occurred and may occur due to a variety of factors, many of which are outside of our control, including, but not limited to:
•our dependence on single source and sole source suppliers for some of the components and materials used in our products;
•production problems and quality issues with the materials we purchase for manufacturing, which could impact our ability to manufacture and ship our products and related components;
•the level of demand for our products, which may vary significantly and result in excess capacity expenses, and our ability to increase penetration in our existing markets and expand into new markets;
•the timing and cost of, and level of investment in, research and development and commercialization activities relating to our products, which may change from time to time;
•the volume and mix of our product and services sales or changes in the manufacturing or sales costs related to our products and services;
•the success of our recently introduced products, including our Explore, Target, Focus and Insight product lines, and the introduction of our own qPCR readout platform, Olink Signature Q100, or others in our industry. The latest addition to Explore offering, Explore HT, was launched mid 2023.
•reductions in capacity or shutdowns of laboratories and other institutions as well as other impacts stemming from the COVID-19 pandemic that may continue to linger, including reduced or delayed spending on products and services as a result of such shutdowns and delays before re-opened laboratories and institutions resume previous levels of research activities that require new purchases of our products and services;
•disruptions in customers’ ongoing experiments or interruptions in the ability of our customers to complete research projects as a result of lingering impacts of the COVID-19 pandemic;
•the timing and amount of expenditures that we may incur to acquire, develop or commercialize additional products and technologies or for other purposes, such as the expansion of our facilities;
•changes in governmental funding of life sciences research and development or changes that impact budgets, budget cycles or seasonal spending patterns of our customers;
/20
•increased inflation and interest rates;
•future accounting pronouncements or changes in our accounting policies;
•the outcome of any future litigation or governmental investigations involving us, our industry or both;
•difficulties encountered in delivering our products and services, whether as a result of external factors such as weather or internal issues such as labor disputes;
•general market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors;
•higher than anticipated warranty costs;
•customers accelerating, canceling, reducing or delaying orders as a result of developments related to litigation;
•the impacts of infectious disease, epidemics, pandemics and outbreaks, including the effects of the COVID-19 pandemic, on our business operations and on the business operations of our customers, manufacturers and suppliers;
•seasonality of customer demand throughout the calendar year;
•the risks of recession; and
•the other factors described in this “Risk Factors” section.
The cumulative effects of the factors discussed above could result in large fluctuations and unpredictability in our quarterly and annual operating results. As a result, comparing our operating results on a period-to- period basis may not be meaningful. Investors should not rely on our past results as an indication of our future performance.
This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any guidance we may provide, or if the guidance we provide is below the expectations of analysts or investors, the price of our common shares and ADSs could decline substantially. Such a price decline could occur even when we have met or exceeded any previously publicly stated guidance we may provide. Our failure to reinstate or provide updated annual revenue guidance in the future may make it more difficult for financial analysts and other investors to value our common shares and ADSs and may result in increased volatility in the price of our common shares and ADSs.
Seasonality causes fluctuations in our revenue and results of operations.
We operate on a December 31st year end and there are significant seasonal factors that cause sales of our products, such as our Explore, Target and Focus product lines, to vary on a quarterly or yearly basis and increase the magnitude of quarterly or annual fluctuations in our operating results. We believe that this seasonality results from a number of factors, including the procurement and budgeting cycles of many of our customers, especially government- or grant-funded customers, whose cycles often coincide with government fiscal year ends. For example, the U.S. government’s fiscal year end occurs in our third quarter and may result in increased sales of our products during such quarter if government-funded customers have unused funds that may be forfeited, or future budgets that may be reduced, if such funds remain unspent at such fiscal year end. Furthermore, the academic budgetary cycle similarly requires grantees to ‘use or lose’ their grant funding, which seems to be tied disproportionately to the end of the calendar year, driving sales higher during the fourth quarter. Similarly, our biopharmaceutical customers typically have calendar year fiscal years which also result in a disproportionate amount of their purchasing activity occurring during our fourth quarter. These factors have contributed, and we expect will continue to contribute in the future, to substantial fluctuations in our quarterly operating results. Because of these fluctuations, it is possible that in some quarters our operating results will fall below the expectations of securities analysts or investors. If that happens, the market price of the ADSs would likely decrease. These fluctuations, among other factors, also mean that our operating results in any particular period may not be relied upon as an indication of future performance. Seasonal or cyclical
/21
variations in our sales have in the past, and may in the future, become more or less pronounced over time, and have in the past materially affected, and may in the future materially affect, our business, financial condition, results of operations and prospects.
Additionally, impacts of the COVID-19 pandemic has caused and may continue to cause unpredictable temporary or permanent fluctuations in seasonal or cyclical variations.
Our sales cycle is lengthy and variable, which makes it difficult for us to forecast revenue and other operating results.
The sales cycle for our products is lengthy and variable because each sale generally represents a major capital expenditure and generally requires the approval of our customers’ senior management. This may contribute to substantial fluctuations in our quarterly or annual operating results, particularly during the periods in which our sales volume is low. Factors that may cause fluctuations in our quarterly or operating results include, without limitation, market acceptance for our new products; our ability to attract new customers; publications of studies by us, competitors or third parties; the timing and success of new product introductions by us or our competitors or other changes in the competitive dynamics of our industry, such as consolidation; the amount and timing of our costs and expenses; changes in our pricing policies or those of our competitors; general economic, industry and market conditions; the effects of seasonality; the regulatory environment; expenses associated with warranty costs or unforeseen product quality issues; the hiring, training and retention of key employees, including our ability to grow our sales organization; litigation or other claims against us for intellectual property infringement or otherwise; our ability to obtain additional financing as necessary; changes or trends in new technologies and industry standards; and the impact of COVID-19. Because of these fluctuations, it is likely that in some future quarters our operating results will fall below the expectations of securities analysts or investors. If that happens, the market price of the ADSs would likely decrease. Such fluctuations also mean that investors may not be able to rely on our operating results in any particular period as an indication of future performance. Sales to existing customers and the establishment of a business relationship with other potential customers is a lengthy process, generally taking several months and sometimes longer. Following the establishment of the relationship, the negotiation of purchase terms can be time-consuming, and a potential customer may require an extended evaluation and testing period. In anticipation of product orders, we may incur substantial costs before the sales cycle is complete and before we receive any customer payments. As a result, in the event that a sale is not completed or is canceled or delayed, we may have incurred substantial expenses, making it more difficult for us to become profitable or otherwise negatively impacting our financial results. Furthermore, because of our lengthy sales cycle, the realization of revenue from our selling efforts may be substantially delayed, our ability to forecast our future revenue may be more limited and our revenue may fluctuate significantly from quarter to quarter.
We may incur impairment charges on our goodwill and intangible assets which could adversely impact our financial results.
Goodwill and certain intangible assets with indefinite lives are tested for impairment annually, or upon the identification of any impairment indicators. As of December 31, 2023, goodwill and intangible assets with indefinite lives represented approximately 35% of our total assets. In the future, if we determine that there has been impairment, our net profit or net loss for the relevant period would be reduced by the amount of the impairment, net of tax effects, if any.
We are exposed to risks related to currency exchange rates.
Due to the international scope of our operations, our assets, earnings and cash flows are affected by fluctuations in the exchange rates of several currencies, particularly the Swedish Kronor (SEK), the Japanese Yen (JPY), the Euro (EUR), the British Pound (GBP), and the Chinese Yuan (CNY). Currency risks arise when future commercial transactions or reported assets or liabilities are denominated in a currency other than our reporting currency, the USD. Exchange rate fluctuations between local currencies and the USD create risk in several ways, including the following:
•weakening of the USD may increase the USD cost of overseas research and development expenses and the cost of sourced product components outside the United States;
•the exchange rates on non-USD transactions and cash deposits can distort our financial results; and
/22
•the pricing and profit margins of our products may be affected by currency fluctuations.
In addition, to the extent our need for contract manufacturing increases once certain of our products reach the commercial market, our exposure to currency risks will increase proportionally. We do not engage in regular hedging transactions, since to date our currency exposure has been mostly related to purchased services for product development, which has been irregular and difficult to anticipate. It is possible that fluctuations in currency exchange rates could have a material adverse effect on our business, results of operations and financial condition.
We are subject to risks related to taxation in multiple jurisdictions.
We are subject to income taxes in Swedish and foreign jurisdictions. Significant judgments based on interpretations of existing tax laws or regulations may be required in determining our provision for income taxes. Our effective income tax rate could be adversely affected by various factors, including, but not limited to, changes in the mix of earnings in tax jurisdictions with different statutory tax rates, changes in the valuation of deferred tax assets and liabilities, changes in existing tax policies, laws, regulations or rates, changes in the level of non-deductible expenses (including share-based compensation), changes in the location of our operations, changes in our future levels of research and development spending, mergers and acquisitions or the result of examinations by various tax authorities. Although we believe our tax estimates are reasonable, if the U.S. Internal Revenue Service (IRS) or other taxing authority disagrees with the positions taken on our tax returns, we could have additional tax liability, including interest and penalties. If material, payment of such additional amounts upon final adjudication of any disputes could have a material impact on our results of operations and financial position.
Changes in tax laws or regulations that are applied adversely to us or our customers may have a material adverse effect on our business, cash flow, financial condition or results of operations.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could affect the tax treatment of our domestic and foreign earnings. Any new taxes could adversely affect our domestic and international business operations and our business and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely and retroactively to us. We will continue to monitor and assess the impact of the tax legislation on our business. Any changes in tax laws or regulations that are applied adversely to us or our customers could have a material adverse effect on our business, cash flow, financial condition or results of operations.
Risks Related to Our Dependence on Third Parties
We are dependent on single source and sole source suppliers for some of the components and materials used in our products and the loss of any of these suppliers could harm our business. The ability of our suppliers to meet our needs and the needs of our customers could be reduced or eliminated by any lingering impacts of the COVID-19 pandemic.
In certain cases, we rely on single source suppliers for all of our requirements for some of our materials or components. In several cases, we do not have long term contracts with these suppliers, and even in the cases where we do, the contracts include significant qualifications that would make it extremely difficult for us to force the supplier to provide us with their services, materials or components should they choose not to do so or do not have the capacity to do so. We are therefore subject to the risk that these third-party suppliers will not be able or willing to continue to provide us with materials and components that meet our specifications, quality standards and delivery schedules. Factors that could impact our suppliers’ willingness and ability to continue to provide us with the required materials and components include disruption at or affecting our suppliers’ facilities, such as work stoppages or natural disasters, infectious disease, epidemics or pandemics, including COVID-19, outbreaks, adverse weather or other conditions that affect their supply, the financial condition of our suppliers, deterioration in our relationships with these suppliers or the decision by such suppliers to introduce products that compete directly with our solutions. In addition, we cannot be sure that we will be able to obtain these materials and components on satisfactory terms. Any increase in material and component costs or decrease in availability could reduce our sales and harm our gross margins. In addition, any loss of a material supplier may permanently cause a change in one or more of our products that may not be accepted by our customers or cause us to eliminate that product altogether.
/23
For example, we depend on a single-source supplier for antibodies used for some of our products. We also depend on single source suppliers for instrumentation used for our products. Lead times for some of these antibodies and instruments can be several months or more and could be exacerbated due to the COVID-19 pandemic. In the event that demand increases, a manufacturing ‘lot’ does not meet our specifications or we fail to forecast and place purchase orders sufficiently in advance, this could result in a material shortage. Some of the antibodies and both of the platforms are proprietary to these suppliers, thereby making second sourcing and development of a replacement difficult. Furthermore, these suppliers have intellectual property rights that could prevent us from sourcing such antibodies and instruments from other suppliers. These suppliers could choose to create products that directly compete with our products and end our current supplier-customer relationships. If antibodies or instruments become unavailable from our current suppliers and we are unable to find acceptable substitutes for these suppliers, we may be required to produce them internally or change our product designs.
We have not qualified secondary sources for all materials or components that we source through a single supplier and we cannot assure investors that the qualification of a secondary supplier will prevent future supply issues. Disruption in the supply of materials or components would impair our ability to sell our products and meet customer demand, and also could delay the launch of new products, any of which could harm our business and results of operations. If we were to have to change suppliers, the new supplier may not be able to provide us with materials or components in a timely manner and in adequate quantities that are consistent with our quality standards and on satisfactory pricing terms. In addition, alternative sources of supply may not be available for materials that are scarce or components for which there are a limited number of suppliers.
While we have taken steps to mitigate supply chain and transportation infrastructure system issues which resulted from the COVID-19 pandemic, the continued impacts of the COVID-19 pandemic, including interruptions in or failures of the global supply chain and transportation infrastructure system, could cause certain of our suppliers to experience shortages in materials and components that we depend on such suppliers to provide, could result in price increases in the materials and components we source from suppliers or could reduce the ability of our suppliers to meet our needs or the needs of our customers. The residual impacts of the COVID-19 pandemic could cause certain of our suppliers to be unable to operate temporarily or go out of business permanently. The realization of any of these risks could prevent us from producing, selling or delivering our products, reduce our sales and harm our gross margins or permanently cause a change in one or more of our products that may not be accepted by our customers or cause us to eliminate that product altogether.
We rely on contract manufacturers for the development and manufacturing of our Olink Signature platform, which can create supply uncertainties.
We rely on contract manufacturers for the production of our Olink Signature platform and, if it proves difficult for contract manufacturers to scale up production of the platform, full-scale production may be delayed.
Our reliance on a third-party service provider for provision of our services in China could limit or prevent us from providing our services and impact our revenue.
We offer Analysis Service through a third-party service provider in China. The ability of our third-party service provider to provide our services has been impacted by the COVID-19 pandemic and may be subject to future disruption. If this third- party service provider does not perform adequately, we may not realize long-term revenue growth in China.
If our third-party providers fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business.
Our third-party manufacturers are subject to numerous environmental, health and safety laws and regulations, including those governing the handling, use, storage, treatment and disposal of hazardous materials and wastes. Although we believe that the safety procedures utilized by our third-party manufacturers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Although we maintain workers’ compensation insurance to cover us for costs and expenses, we may incur due to injuries resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate
/24
coverage against potential liabilities. We do not carry specific biological waste or hazardous waste insurance coverage, workers’ compensation or property and casualty and general liability insurance policies that include coverage for damages and fines arising from biological or hazardous waste exposure or contamination.
Risks Related to Intellectual Property
If we are unable to protect our intellectual property effectively, our business would be harmed.
We rely on patent protection as well as trademark, copyright, trade secret and other intellectual property rights protection and contractual restrictions to protect our proprietary technologies, all of which provide limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. As of December 31, 2023, worldwide we owned or in-licensed 44 issued or allowed patents across ten patent families (of which 22 patents are national validations of granted European patents, corresponding to six granted European patents each validated in three or four European countries) and 47 pending patent applications across nine patent families (of which four applications are still in the priority year). Although we keep other aspects of our proprietary technologies as trade secrets, we cannot assure investors that we will keep our competitive advantage against third parties after the expiration of these patent families. We continue to file new patent applications to attempt to obtain further legal protection of the full range of our technologies. If we fail to protect our intellectual property, third parties may be able to compete more effectively against us and we may incur substantial litigation costs in our attempts to recover or restrict the use of our intellectual property.
Our success depends in part on obtaining patent protection for our products and services, preserving trade secrets, patents, copyrights and trademarks, operating without infringing the proprietary rights of third parties and acquiring licenses for technology or products. We may exercise our business judgment and choose to relinquish rights in trade secrets by filing applications that disclose and describe our inventions and certain trade secrets when we seek patent protection for certain of our products and technology. We cannot assure investors that any of our currently pending or future patent applications will result in issued patents and we cannot predict how long it will take for such patents to be issued. Further, we regularly file patent applications on certain aspects of our products and technologies in a single nation (e.g. United Kingdom) or region (e.g. the European Patent Office) in order to obtain a priority date for these aspects of our products and technologies. Such national or regional patent applications are not eligible to become an issued patent outside of the respective nation or region until, among other things, we file an international patent application or other foreign applications within 12 months of the filing date of the earlier patent application. Such applications may not become issued patents for a variety of reasons, including our failure to file international applications or other foreign applications within the permitted timeframe or a decision that doing so no longer makes business or financial sense. Publications of discoveries in scientific literature often lag behind the actual discoveries and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain, despite the importance of seeking patent protection in our industry. Our pending and future patent applications may not result in patents being issued that protect our product candidates, in whole or in part, or which effectively prevent others from commercializing competitive product candidates. Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative product candidates in a non-infringing manner.
Further, we cannot assure investors that other parties will not challenge any patents issued to us or that courts or regulatory agencies will hold our patents to be valid or enforceable. We cannot guarantee investors that we will be successful in defending challenges made against our patents and patent applications, even if we spend significant resources defending such challenges. Any successful third-party challenge to our patents could result in the unenforceability or invalidity of such patents and could deprive us of the ability to prevent others from using the technologies claimed in such issued patents. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
Changes in either the patent laws or in interpretations of patent laws in the United States or other jurisdictions may diminish the value of our intellectual property. We cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents.
In addition to pursuing patents on our technology, we take steps to protect our intellectual property and proprietary technology by entering into confidentiality agreements and intellectual property assignment agreements with our employees, consultants, corporate partners and, when needed, our advisors. Such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements and we may not be able to prevent such unauthorized disclosure. Monitoring unauthorized disclosure is difficult and we do not know whether the steps we have taken to prevent such
/25
disclosure are, or will be, adequate. If we were to enforce a claim that a third-party had illegally obtained and was using our trade secrets, it would be expensive and time consuming and the outcome would be unpredictable.
With respect to all categories of intellectual property protection, our competitors could purchase our products and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. In addition, competitors may develop their own versions of our products in countries where we did not apply for patents, where our patents have not issued or where our intellectual property rights are not recognized and compete with us in those countries and markets.
The laws of some countries do not protect intellectual property rights to the same extent as the laws of the United States and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biotechnology, which could make it difficult for us to stop the infringement of our patents. The legal systems in certain countries may also favor state-sponsored or companies headquartered in particular jurisdictions over our first-in-time patents and other intellectual property protection. We are aware of incidents where such entities have stolen the intellectual property of domestic companies in order to create competing products and we believe we may face such circumstances ourselves in the future. In the USTR annual “Special 301” Report released in 2019, the adequacy and effectiveness of intellectual property protection in a number of foreign countries were analyzed. A number of countries in which both we and our distributors operate are identified in the report as being on the Priority Watch List. In China, for instance, the USTR noted a range of IP-related concerns, including a need to “strengthen IP protection and enforcement, including as to trade secret theft, online piracy and counterfeiting, the high- volume manufacture and export of counterfeit goods, and impediments to pharmaceutical innovation.” The absence of harmonized intellectual property protection laws and effective enforcement makes it difficult to ensure consistent respect for patent, trade secret, and other intellectual property rights on a worldwide basis. As a result, it is possible that we will not be able to enforce our rights against third parties that misappropriate our proprietary technology in those countries.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents, trademarks, copyrights or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming and divert the time and attention of our management and scientific personnel. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents, in addition to counterclaims asserting that our patents are invalid or unenforceable, or both. In any patent infringement proceeding, there is a risk that a court will decide that a patent of ours is invalid or unenforceable, in whole or in part, and that we do not have the right to stop the other party from using the invention at issue. There is also a risk that, even if the validity of such patents is upheld, the court will construe the patent’s claims narrowly or decide that we do not have the right to stop the other party from using the invention at issue on the grounds that our patent claims do not cover the invention. An adverse outcome in a litigation or proceeding involving our patent could limit our ability to assert those patents against those parties or other competitors and may curtail or preclude our ability to exclude third parties from making and selling similar or competitive products. Similarly, if we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the trademarks in question. In this case, we could ultimately be forced to cease use of such trademarks.
Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could adversely affect the price of our common shares and ADSs. Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. Even if we ultimately prevail in such claims, the monetary cost of such litigation and the diversion of the attention of our management and scientific personnel could outweigh any benefit we receive as a result of the proceedings.
Additionally, for certain of our existing and future in-licensed patent rights, we may not have the right to bring suit for infringement and may have to rely on third parties to enforce these rights for us. If we cannot or choose not to take action against those, we believe infringe our intellectual property rights, we may have difficulty competing in certain markets where such potential infringers conduct their business, and our commercialization efforts may suffer as a result.
Issued patents covering our products and services could be found invalid or unenforceable if challenged.
/26
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability and some of our patents or patent applications, including licensed patents, may be challenged in courts or patent offices in the United States and abroad in opposition, derivation, reexamination, inter partes review, post- grant review or interference. Additionally, if we and our licensing partners initiate or become involved in legal proceedings against a third party to enforce a patent covering one of our products or technologies, the defendant could counterclaim that the patent covering our product is invalid or unenforceable. In patent litigation in the United States, counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including patent eligible subject matter, lack of novelty, obviousness or non- enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the United States Patent and Trademark Office (USPTO), or made a misleading statement, during prosecution. In addition, the United States now awards patent priority to the first party to file a patent application, and others may submit patent claims covering our inventions prior to us. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. A successful third-party challenge to our patents could result in the unenforceability or invalidity of such patents, which could have a material adverse impact on our business. Furthermore, if the breadth or strength of protection provided by our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future products and services.
We may not be aware of all third-party intellectual property rights potentially relating to our platforms, products and services. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until approximately 18 months after filing or, in some cases, not until such patent applications issue as patents. We might not have been the first to make the inventions covered by each of our pending patent applications and we might not have been the first to file patent applications for these inventions. To determine the priority of these inventions, we may have to participate in interference proceedings, derivation proceedings or other post-grant proceedings declared by the USPTO. The outcome of such proceedings is uncertain, and other patent applications may have priority over our patent applications. Such proceedings could also result in substantial costs to us and divert our management’s attention and resources.
We may not be able to protect and enforce our trademarks.
We have not yet registered certain of our trademarks in all of our potential markets, although we have registered the trademark PROSEEK in the European Union and China and the trademarks OLINK,  OLINK and in the European Union, United States, Canada, China, United Kingdom, Japan, Norway, Singapore and a number of other countries. As we apply to register our as yet unregistered trademarks in the United States and other countries, our applications may not be allowed for registration in a timely fashion or at all, and our registered trademarks may not be maintained or enforced. In addition, opposition or cancellation proceedings may be filed against our trademark applications and registrations, and our trademarks may not survive such proceedings. In certain countries outside of the United States, trademark registration is required to enforce trademark rights. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
OLINK and in the European Union, United States, Canada, China, United Kingdom, Japan, Norway, Singapore and a number of other countries. As we apply to register our as yet unregistered trademarks in the United States and other countries, our applications may not be allowed for registration in a timely fashion or at all, and our registered trademarks may not be maintained or enforced. In addition, opposition or cancellation proceedings may be filed against our trademark applications and registrations, and our trademarks may not survive such proceedings. In certain countries outside of the United States, trademark registration is required to enforce trademark rights. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
OLINK and in the European Union, United States, Canada, China, United Kingdom, Japan, Norway, Singapore and a number of other countries. As we apply to register our as yet unregistered trademarks in the United States and other countries, our applications may not be allowed for registration in a timely fashion or at all, and our registered trademarks may not be maintained or enforced. In addition, opposition or cancellation proceedings may be filed against our trademark applications and registrations, and our trademarks may not survive such proceedings. In certain countries outside of the United States, trademark registration is required to enforce trademark rights. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.If we are sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and could prevent or delay us from developing or commercializing our products.
Our commercial success depends, in part, on our ability to develop, manufacture, market and sell our products and future product candidates without infringing the intellectual property and other proprietary rights of third parties. However, our development and commercialization activities may be subject to claims that we infringe or otherwise violate patents or other intellectual property rights owned or controlled by third parties. Third parties may have United States and non-U.S. issued patents and pending patent applications relating to compounds, methods of manufacturing compounds and/or methods of use for the applications for which we are developing our product candidates. If any third-party patents or patent applications are found to cover our product candidates or their methods of use or manufacture, we may not be free to manufacture or market our product candidates as planned without obtaining a license, which may not be available on commercially reasonable terms or at all, or it may be non-exclusive, which could result in our competitors gaining access to the same intellectual property.
There is a substantial amount of intellectual property litigation in the life sciences industry, and we may become party to, or threatened with, litigation or other adversarial proceedings regarding intellectual property rights with respect to our products and products candidates, including patent infringement lawsuits in Europe, the United States or abroad, as well as interference, derivation, inter partes review, and post-grant proceedings before the European Patent Office (EPO) or USPTO and opposition or other proceedings before foreign patent offices. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the composition, use or manufacture of our products and product candidates. We cannot guarantee that any of our patent searches or analyses including, but not limited to, the identification of relevant patents, the scope of patent claims or the expiration of relevant patents are complete or thorough, nor can we be certain that we have identified each and every
/27
patent and pending application in the United States, Europe and other jurisdictions that is relevant to or necessary for the commercialization of our product candidates in any jurisdiction. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our product candidates may be accused of infringing. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Accordingly, third parties may assert infringement claims against us based on intellectual property rights that exist now or arise in the future. The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The life sciences industry has produced a significant number of patents, and it may not always be clear to industry participants, including us, which patents cover various types of products or methods of use or manufacture. The scope of protection afforded by a patent is subject to interpretation by the courts, and the interpretation is not always uniform. If we were sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could significantly harm our business and operating results. In addition, parties making claims against us may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources, and we may not have sufficient resources to bring these actions to a successful conclusion. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure.
If we are found to infringe a third party’s intellectual property rights, we could be forced, including by court order, to cease developing, manufacturing or commercializing the infringing product candidate or product. Alternatively, we may be required to obtain a license from such third party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing product candidate or product. If we were required to obtain a license to continue to manufacture or market the affected product, we may be required to pay substantial royalties or grant cross-licenses to our patents. We cannot, however, be certain that any such license will be available on acceptable terms, if at all. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations as a result of claims of patent infringement or violation of other intellectual property rights. Further, the outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance, including the demeanor and credibility of witnesses and the identity of any adverse party. This is especially true in intellectual property cases that may turn on the testimony of experts as to technical facts upon which experts may reasonably disagree. Furthermore, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us; alternatively or additionally it could include terms that impede or destroy our ability to compete successfully in the commercial marketplace. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise additional funds or otherwise have a material adverse effect on our business, results of operations, financial condition and prospects.
Obtaining and maintaining our patent protection depends on compliance with various procedural, documentary, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with any of these requirements.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO and national patent offices in several stages over the lifetime of the patent. The USPTO, the EPO and various foreign governmental patent offices require compliance with a number of procedural, documentaries, fee payment (including annuities) and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors or collaboration partners fail to maintain the patents and patent applications covering our product candidates, our competitors might be able to enter the market, which would have an adverse effect on our business.
We may not be able to protect our intellectual property rights throughout the world.
/28
Filing, prosecuting and defending patents on product and product candidates throughout the world is prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biotechnology, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Patent terms may be inadequate to protect our competitive position on our products and services for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest United States non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our products and services are obtained, once the patent life has expired, we may be open to competition from competitive products. Given the amount of time required for the development, testing and regulatory review of new products and services, patents protecting such products and services might expire before or shortly after such products and services are commercialized.
As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biotechnology companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involve both technological complexity and legal complexity. Therefore, obtaining and enforcing biotechnological patents is costly, time-consuming and inherently uncertain. In addition, the America Invents Act (AIA) has been enacted in the United States, resulting in significant changes to the United States patent system.
An important change introduced by the AIA is that, as of March 16, 2013, the United States transitioned to a “first-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. A third party that files a patent application in the USPTO after that date but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by the third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application, but circumstances could prevent us from promptly filing patent applications on our inventions.
Among some of the other changes introduced by the AIA are changes that limit where a patentee may file a patent infringement suit and that provide opportunities for third parties to challenge any issued patent in the USPTO. This applies to all of our United States patents, even those issued before March 16, 2013.
Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. The AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Additionally, the United States Supreme Court and the Court of Appeals for the Federal Circuit have ruled on patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations, especially with regards to certain inventions or discoveries relating to the life sciences. For example, certain decisions stand for the proposition that patent claims that recite laws of nature (for example, the relationships between the levels of certain biomarkers and the likelihood of risk of recurrence of cancer) are not themselves patentable unless those patent claims have sufficient additional features that provide practical assurance
/29
that the processes are genuine inventive applications of those laws rather than patent drafting efforts designed to monopolize the law of nature itself. What constitutes a “sufficient” additional feature is uncertain. Furthermore, in view of these decisions, in December 2014 the USPTO published revised guidelines for patent examiners to apply when examining process claims for patent eligibility. This guidance has been periodically updated by the USPTO since 2014, most recently in 2019. The guidance indicates that claims directed to a law of nature, a natural phenomenon or an abstract idea that do not meet the eligibility requirements should be rejected as non-statutory, patent ineligible subject matter; however, method of treatment claims that practically apply natural relationships should be considered patent eligible. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the United States Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We or our licensors may be subject to claims that former employees, collaborators or other third parties have an interest in our owned or in-licensed patents, trade secrets, or other intellectual property as an inventor or co-inventor. For example, we or our licensors may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or our or our licensors’ ownership of our owned or in-licensed patents, trade secrets or other intellectual property. If we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property that is important to our product candidates. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
Some of the intellectual property that is important to our business is owned by other companies or institutions and licensed to us, and changes to the rights we have licensed may adversely impact our business.
We license from third parties some of the intellectual property that is important to our business and may need to obtain additional licenses from others to advance our research and development or commercialization activities. Our license agreements, and we expect that future license agreements will impose, various development, diligence, commercialization, and other obligations on us. If we fail to meet our obligations under these licenses, or if we have a dispute regarding the terms of the licenses, these third parties could terminate the licenses. If the third parties who license intellectual property to us fail to maintain the intellectual property that we have licensed, or lose rights to that intellectual property, the rights we have licensed may be reduced or eliminated, which could subject us to claims of intellectual property infringement. Termination of these licenses or reduction or elimination of our licensed rights may result in our having to negotiate new or reinstated licenses with less favorable terms or could subject us to claims of intellectual property infringement or contract breach in litigation or other administrative proceedings that could result in damage awards against us and injunctions that could prohibit us from selling our products. We may incur increased costs to replace such licenses and it may take a few months to find suitable replacements.
In addition, some of our licenses from third parties limit the field in which we can use the licensed technology. Therefore, in order for us to use such licensed technology in potential future applications that are outside the licensed field of use, we may be required to negotiate new licenses with our licensors or expand our rights under our existing licenses. We cannot assure you that we will be able to obtain such licenses or expanded rights on reasonable terms or at all.
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including: the scope of rights granted under the license agreement and other interpretation-related issues; the extent to which our product candidates, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing
/30
agreement; the sublicensing of patent and other rights under our collaborative development relationships; our diligence obligations under the license agreement and what activities satisfy those diligence obligations; the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and the priority of invention of patented technology. In the event a dispute with our licensors were to occur, our licensors may seek to renegotiate the terms of our licenses, increase the royalty rates that we pay to obtain and maintain those licenses, limit the field or scope of the licenses, or terminate the license agreements. Further, because of the rapid pace of technological change in our industry, we may need to rely on key technologies developed or licensed by third parties, and we may not be able to obtain licenses and technologies from these third parties at all or on reasonable terms. The occurrence of these events may have a material adverse effect on our business, financial condition or results of operations.
Confidentiality and non-compete agreements with employees and others may not adequately prevent disclosure of trade secrets and protect other proprietary information.
We consider proprietary trade secrets, confidential know-how and unpatented know-how to be important to our business. We may rely on trade secrets or confidential know-how to protect our technology, especially where patent protection is believed to be of limited value. However, trade secrets and confidential know-how are difficult to maintain as confidential.
To protect this type of information against disclosure or appropriation by competitors, our policy is to require our employees, consultants, contractors and advisors to enter into confidentiality agreements with us, in addition to agreements with covenants not to compete or solicit employees or customers.
However, current or former employees, consultants, contractors and advisers may unintentionally or willfully disclose our confidential information to competitors or otherwise compete, and confidentiality agreements and covenants not to compete or solicit may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party obtained illegally and is using trade secrets or confidential know-how, or is wrongfully engaging former employees and consultants in breach of their contracts with us, is expensive, time-consuming and unpredictable. The enforceability of confidentiality agreements and covenants not to compete or solicit may vary from jurisdiction to jurisdiction. Furthermore, if a competitor lawfully obtained or independently developed any of our trade secrets, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret.
Failure to obtain or maintain trade secrets or confidential know-how trade protection could adversely affect our competitive position. Moreover, our competitors may independently develop substantially equivalent proprietary information and may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, our competitors could limit our use of our trade secrets or confidential know-how.
Under certain circumstances, we may also decide to publish some know-how to attempt to prevent others from obtaining patent rights covering such know-how.
Failure or a breach of our information technology systems, loss of data and other disruptions could adversely affect our business and our reputation and expose us to liability.
Our ability to execute our business plan and to comply with regulatory requirements with respect to data control and data integrity depends, in part, on the continued and uninterrupted performance of our information technology systems. These systems are vulnerable to damage due to a variety of factors, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. We will continue to update policies and procedures to provide protections against such problems in the future and have purchased cybersecurity insurance, although such insurance may not be sufficient to cover us for any losses or damages, we may face. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our IT systems, there are no assurances that electronic break-ins, computer viruses and similar disruptive problems, and/or sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data will not occur. The occurrence of any of the foregoing with respect to our IT systems could have a material adverse effect on our business, results of operations or financial condition.
In the ordinary course of our business, we and our collaborators collect and store sensitive data, intellectual property and proprietary business information owned or controlled by ourselves or our customers, our collaborators, government entities and other parties. We manage and maintain our applications and data through a combination of on-site systems and cloud- based data centers. We utilize external security and infrastructure vendors to manage components of our data centers. We face a number of risks related to protecting this sensitive information, including loss-of-access risk, unauthorized access, use, disclosure or modification, and the risk of our inability to adequately monitor, audit and modify
/31
our respective control over our critical information. This risk extends to the data we entrust to the third-party vendors and subcontractors that help us manage this sensitive data or otherwise process it on our behalf.
The secure processing, storage, maintenance and transmission of this sensitive information are vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take reasonable measures to protect sensitive and proprietary data from unauthorized access, use or disclosure, no security measures can be perfect and our respective information technology and infrastructure may be vulnerable to attacks by hackers or malicious software or breached due to employee error, malfeasance or other malicious or inadvertent disruptions (including actions or inactions by those with authorized access to our networks). Any such breach or interruption could compromise our networks and the information stored there could be accessed by unauthorized parties, publicly disclosed, lost or stolen. Any such access, breach or other loss of information could result in legal claims or proceedings, liability under our customer contracts or federal or state laws that protect the privacy of personal information and regulatory penalties. Notice of breaches may be required to be provided to affected individuals, federal, state and foreign regulators, the media or state attorneys general. Such a notice could harm our reputation and ability to compete. Although we have implemented security measures and formal, dedicated enterprise security programs to prevent unauthorized access to personal data, such data is currently accessible through multiple channels and we may experience one or more data breaches. We have adopted and will continue to update policies and procedures to provide protections against such attacks in the future and have purchased cybersecurity insurance as protection in the future. Despite the precautionary measures we have taken to prevent unanticipated problems, additional attacks may occur in the future. Unauthorized access, loss or dissemination could also disrupt our operations and damage our reputation, which could adversely affect our results of operations and financial condition. Our insurance policies may not be adequate to compensate us for the potential losses arising from any such disruption in or, failure or security breach of our systems or third-party systems where information important to our business operations or commercial development is stored. In addition, such insurance may not be available to us in the future on economically reasonable terms, or at all. Further, our insurance may not cover all claims made against us and could have high deductibles in any event, and defending a suit, regardless of its merit, could be costly and divert management attention.
Furthermore, our contractors and consultants are vulnerable to damage from computer viruses and unauthorized access. We rely on a few third parties for the provision of subcontracted Analysis Services, as well as administrative services, and security breaches, loss of data and other disruptions relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our products could be delayed.
Risks Related to Our Employee Matters, Managing Our Growth and Other Risks Relating to Our Operations
We will continue to develop and expand our workforce and commercial infrastructure to support anticipated growth and scaling up in demand for our products and services, and we may encounter difficulties in managing this development and expansion and in meeting fluctuations in this demand.
We will continue to expand our workforce and commercial infrastructure to support anticipated growth and scaling up in demand for our products and services. If we are unable to support fluctuations in the demand for our products and services, including ensuring that we have adequate capacity to meet increased demand, our business could suffer. As of December 31, 2023, we had 707 full-time employees and we will continue to invest and expand our organization as needed to support our growth potential and strategy. We also may expand the scope of our operations as we continue to develop our products and services. As we and our collaborators commercialize additional products and services, we may need to incorporate new equipment, implement new technology systems and laboratory processes and hire new personnel with different qualifications. Failure to manage this growth or transition could result in turnaround time delays, higher service costs, declining service quality, deteriorating customer service and slower responses to competitive challenges. A failure in any one of these areas could make it difficult for us to meet market expectations for our products and services and could damage our reputation and the prospects for our business. Furthermore, the decline in the supply of labor as a result of the impacts of COVID-19 pandemic as well as the current surge in demand for labor and rising labor wages have created labor shortages and higher labor costs. These factors may increase our costs and negatively impact our ability to attract and retain qualified employees.
To manage our continued expansion, we must continue to implement and improve our managerial, operational and financial systems, continue to expand our facilities (including our corporate headquarters in Uppsala, Sweden and our Analysis Service labs in Waltham, Massachusetts and Uppsala, Sweden) and continue to recruit and train additional qualified personnel. Also, our management team may need to divert a disproportionate amount of its attention away from its day-to-day activities and devote a substantial amount of time to managing these development activities. This may result in weaknesses in our infrastructure, operational mistakes, slower development of our products and services, missed or delayed milestone achievement, significant cost overruns, loss of business opportunities, loss of employees, inability to execute on hiring plans and reduced productivity among remaining employees.
/32
If our management is unable to effectively manage our expected development and expansion, our expenses may increase more than expected, our ability to generate or increase our revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance, and our ability to develop and commercialize our products and services and compete effectively, will depend, in part, on our ability to effectively manage our future development and expansion.
Our future success is dependent upon our ability to further penetrate our existing customer base and attract new customers.
Our current customer base is primarily composed of academic and governmental research institutions, as well as biopharmaceutical and contract research organizations (CROs). Our success will depend upon our ability to respond to the evolving needs of and increase our market share among existing customers and add new customers. Identifying, engaging and marketing to customers requires substantial time, expertise and expense and involves a number of risks, including:
•our ability to attract, retain and manage the sales, marketing and service personnel necessary to increase our customer base and broaden market acceptance for our PEA technology platform and existing product lines;
•the time and cost of maintaining and growing a specialized sales, marketing and service infrastructure; and
•our sales force, marketing and service organization may be unable to successfully execute on our commercial strategy.
We have utilized third parties to assist with sales, distribution and customer support in certain regions of the world. There is no guarantee, when we enter into such arrangements, that we will be successful in attracting desirable sales and distribution partners. There is also no guarantee that we will be able to enter into such arrangements on favorable terms. Any failure of our sales and marketing efforts, or those of any third-party sales and distribution partners, would adversely affect our business.
We do not have long-term contracts with customers and a reduction in orders from a significant number of customers could reduce our sales and harm our operating results.
We do not have long-term contracts with all of our customers, and our customer contracts generally do not contain minimum purchase requirements. Therefore, our sales are subject to changes in demand from our customers. The level and timing of orders placed by our customers vary for a number of reasons, including individual customer strategies, availability of funding, the introduction of new technologies, the desire of our customers to reduce their exposure to any single supplier and general economic conditions. In addition, though we believe customers in our markets display a significant amount of loyalty to a particular product, we may not be able to renew a contract on favorable pricing terms if our competitors reduce their prices in order to procure business, or if a customer insists that we lower the price charged under the contract being renewed in order to retain the contract. In addition, if we enter into a contract with a customer on unfavorable terms, it may harm our ability to negotiate future contracts with that customer or other customers. The loss of sales or the reduced profitability of such sales could adversely affect our business, financial position and results of operations.
We depend on our key personnel and other highly qualified personnel, and if we are unable to recruit, train, retain and ensure the health and safety of our personnel, we may not achieve our goals.
Our future success depends on our ability to recruit, train, retain and motivate key personnel, including our senior management, research and development, manufacturing and sales, customer service and marketing personnel. Competition for qualified personnel is intense. As we grow, we may continue to make changes to our management team, which could make it difficult to execute on our business plans and strategies. New hires also require significant training and, in most cases, take significant time before they achieve full productivity. Our failure to successfully integrate these key personnel into our business could adversely affect our business.
Our continued growth depends, in part, on attracting, retaining and motivating highly trained sales personnel with the necessary scientific background and ability to understand our systems at a technical level to effectively identify and sell to potential new customers. We also compete for computational biologists and qualified scientific personnel with other life sciences companies, academic institutions and research institutions.
We do not maintain key person life insurance or fixed term employment contracts with any of our employees. As a result, employees, except as prohibited by non-competition provisions or applicable law or regulation, could leave our company with little or no prior notice and would be free to work for a competitor. Because of the complex and technical nature of our products and the dynamic market in which we compete, any failure to attract, train, retain and motivate qualified personnel could materially harm our operating results and growth prospects. Additionally, while we are committed to maintaining a
/33
safe workplace, the health and safety of our personnel may continue to be impacted by COVID-19 and our operating results and growth prospects could be materially harmed as a result.
We are subject to the United States Foreign Corrupt Practices Act and anti-corruption laws of other countries, as well as export control laws, customs laws, sanctions laws and other laws governing our operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures, and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations are subject to certain anti-corruption laws, including the United States Foreign Corrupt Practices Act (FCPA), and other anticorruption laws that apply in countries where we do business. The FCPA and other anti-corruption laws generally prohibit us and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential FCPA violations and we participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the FCPA or local anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We are also subject to other laws and regulations governing our international operations, including regulations administered in the United States and in the European Union, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations (collectively, Trade Control Laws).
There can be no assurance that we will be completely effective in ensuring our compliance with all applicable anticorruption laws, including the FCPA or other legal requirements, such as Trade Control Laws. Any investigation of potential violations of the FCPA, other anti-corruption laws or Trade Control Laws by the United States, the European Union or other authorities could have an adverse impact on our reputation, our business, results of operations and financial condition. Furthermore, should we be found not to be in compliance with the FCPA, other anti-corruption laws or Trade Control Laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, as well as the accompanying legal expenses, any of which could have a material adverse effect on our reputation and liquidity, as well as on our business, results of operations and financial condition.
European data collection is governed by restrictive laws and regulations governing the use, disclosure or other processing and cross-border transfer of personal information.
The collection and use of personal data, including health-related data, in the European Economic Area (EEA) (being the European Union plus Norway, Iceland and Liechtenstein) is governed by the European Union’s General Data Protection Regulation 2016/679 (GDPR), which became effective May 25, 2018, and related applicable data protection and privacy laws of the member states of the EEA and the United Kingdom. The GDPR applies to the processing of personal data by any company established in the EEA and to companies established outside the EEA to the extent they process personal data in connection with the offering of goods or services to data subjects in the EEA or the monitoring of the behavior of data subjects in the EEA. The GDPR is wide-ranging in scope and imposes numerous additional requirements on companies that process personal data of EEA data subjects, including imposing special requirements in respect of the processing of health and other sensitive data. The GDPR enhances data protection obligations for data controllers of personal data, including stringent requirements relating to the consent of data subjects, expanded disclosures about how personal data is used, requirements to conduct data protection impact assessments for “high risk” processing, limitations on retention of personal data, mandatory data breach notification and “privacy by design” requirements, and creates direct obligations on service providers acting as processors. It also establishes rights for individuals with respect to their personal data, including rights of access and deletion in certain circumstances.
The GDPR also imposes strict rules on the transfer of personal data outside of the EEA to countries that do not ensure an adequate level of protection, like the United States (so-called “third countries”). These transfers are prohibited unless an appropriate safeguard specified by the GDPR is implemented, such as the Standard Contractual Clauses (SCCs) approved by the European Commission, or a derogation applies. The Court of Justice of the European Union (CJEU) confirmed in its judgment in the "Schrems II" case (Case C‑311/18) in July 2020 that the SCCs remain a valid mechanism for transfers of personal data to third countries. However, the CJEU also ruled that transfers made pursuant to the SCCs and other alternative transfer mechanisms need to be analyzed on a case-by-case basis to ensure EU standards of data protection are met in the jurisdiction where the data importer is based, and there continue to be concerns about whether the SCCs and other mechanisms will face additional challenges. European regulators have issued recent guidance following the CJEU case that imposes significant new diligence requirements on transferring data outside the EEA, including under an approved transfer mechanism. This guidance requires an “essential equivalency” assessment of the laws of the destination country. If essentially equivalent protections are not available in the destination country, the exporting entity must then assess if supplemental measures can be put in place that, in combination with the chosen transfer mechanism, would address the
/34
deficiency in the laws and ensure that essentially equivalent protection can be given to the data. Complying with this guidance will be expensive and time consuming and may, in the worst case scenario, ultimately prevent us from transferring personal data outside the EEA, which would cause significant business disruption. Like many other businesses, until the legal uncertainties regarding how to legally continue transfers pursuant to the SCCs and other mechanisms are settled, we will continue to face uncertainty as to whether our efforts to comply with our obligations under the GDPR will be sufficient. This and other future developments regarding the flow of data across borders could increase the complexity of transferring personal data across borders in some markets and may lead to governmental enforcement actions, litigation, fines and penalties or adverse publicity, which could have an adverse effect on our reputation and business. That said, as far as transfers of personal data from the EU to the US are concerned, on 10 July 2023, the EU and US announced that they had reached an agreement in principle on a new deal to allow personal data to transfer from the EEA to the US to try and resolve the uncertainty created by the above decision (known as the EU-US Data Privacy Framework, or the "DPF") and to replace the previous EU-US Privacy Shield framework which was invalidated in the "Schrems II" case. Companies participating in the DPF may rely on this framework to receive personal data from the EU/EEA. The US is now subject to an adequacy decision from the European Commission which is now in place. Similar to the EU-US Privacy Shield, the DPF has already been challenged. The challenge is still in its very early stages and the result and potential effects of the challenge is yet unclear. Even if this particular challenge is unsuccessful, it is likely that the DPF will be subject to legal review, with the ultimate risk consisting of the invalidation of the DPF as an EU-US transfer tool.
Failure to comply with the requirements of the GDPR and the related national data protection laws of the European Union Member States and Norway, Iceland and Liechtenstein may result in fines up to €20 million or 4% of a company’s global annual revenues for the preceding financial year, whichever is higher. The authorities have shown a willingness to impose significant fines and issue orders preventing the processing of personal data on non-compliant businesses. Moreover, the GDPR grants data subjects the right to claim material and non-material damages resulting from infringement of the GDPR and introduces the right for non-profit organizations to bring claims on behalf of data subjects. Given the breadth and depth of changes in data protection obligations, maintaining compliance with the GDPR requires significant time, resources and expense, and we may be required to put in place additional controls and processes ensuring compliance as the regulatory landscape continues to evolve. This may be onerous and adversely affect our business, financial condition and results of operations.
Further, the United Kingdom’s vote in favor of exiting the European Union, often referred to as Brexit, and ongoing developments in the United Kingdom means that the EU GDPR regime no longer applies in the United Kingdom, though the substantive law remains very similar. Following the United Kingdom’s withdrawal from the European Union on January 31, 2020, pursuant to the transitional arrangements agreed to between the United Kingdom and European Union, the GDPR continued to have effect in United Kingdom law and continued to do so until December 31, 2020, as if the United Kingdom remained a Member State of the European Union for such purposes. Following December 31, 2020, and the expiry of those transitional arrangements, the data protection obligations of the GDPR continue to apply to United Kingdom-related processing of personal data in substantially unvaried form under the so-called “UK GDPR” (i.e., the GDPR as it continues to form part of law in the United Kingdom by virtue of section 3 of the European Union (Withdrawal) Act 2018, as amended (including by the various Data Protection, Privacy and Electronic Communications (Amendments etc.) (EU Exit) Regulations)). However, going forward, there will be increasing scope for divergence in application, interpretation and enforcement of data protection law as between the United Kingdom and EEA. The government in the United Kingdom has in fact recently proposed a new data protection law (the Data Protection and Digital Information Bill, or "DPDI Bill"); the DPDI Bill is currently some way from being finalized but would, if adopted, make various changes to the existing data protection framework in the United Kingdom, although many aspects of the current data protection regime are likely to remain substantially similar.
Furthermore, the relationship between the United Kingdom and the EEA in relation to certain aspects of data protection law remains somewhat uncertain. For example, with respect to transfers of personal data from the EEA to the United Kingdom, the United Kingdom received an adequacy decision from the EU Commission on 28 June 2021 confirming that, for the time being, the United Kingdom is considered to provide a level of protection for personal data equivalent to that which exists within the EU. This means that, for the moment, transfers of personal data from the EEA to the United Kingdom may continue without any need for additional safeguards (such as EU standard contractual clauses). Importantly, the current adequacy decision for the United Kingdom contains a "sunset clause" which means that it will expire on 27 June 2025, unless the EU Commission decides to renew it at that stage. The current adequacy decision also contains various ongoing monitoring mechanisms, which allow the EU Commission to keep the position under review in the event that there are any future changes to data protection law in the United Kingdom which materially reduce the level of protection provided for personal data. For the time being, however, transfers of personal data from the EEA to the United Kingdom can continue on the basis of the current adequacy decision for the United Kingdom, and no additional safeguards are required. The United Kingdom has also similarly recognized the EEA states as adequate for the purposes of the UK GDPR (under "adequacy regulations", which
/35
are the United Kingdom's equivalent of EU adequacy decisions under the GDPR), meaning that personal data can currently be transferred from the United Kingdom to the EEA without any need for UK standard contractual clauses or other safeguards. Transfers from the UK to the US are also subject to partial UK finding of adequacy provided that the data transferred is covered by what is known as the UK Extension to the new EU-US DPF.
Additionally, as noted above, the United Kingdom has transposed the GDPR into United Kingdom domestic law by way of the UK GDPR with effect from January 2021, which could expose us to two parallel regimes, each of which potentially authorizes similar fines and other potentially divergent enforcement actions for certain violations. Also, following the expiry of the post-Brexit transitional arrangements, the United Kingdom Information Commissioner’s Office is not able to be our “lead supervisory authority” in respect of any “cross border processing” for the purposes of the GDPR. For so long as we are unable to, and/or do not, designate a lead supervisory authority in an EEA member state, with effect from January 1, 2021, we are not able to benefit from the GDPR’s “one stop shop” mechanism. Amongst other things, this would mean that, in the event of a violation of the GDPR affecting data subjects across the United Kingdom and the EEA, we could be investigated by, and ultimately fined by the United Kingdom Information Commissioner’s Office and the supervisory authority in each and every EEA member state where data subjects have been affected by such violation. Other countries have also passed or are considering passing laws requiring local data residency and/or restricting the international transfer of data.
Our business is subject to economic, political, regulatory and other risks associated with international operations.
As a company incorporated and based in Sweden, our business is subject to risks associated with conducting business in Sweden, the European Union, the United States and internationally. Accordingly, our future results could be harmed by a variety of factors, including:
•economic weakness, including inflation, or political instability;
•differing regulatory requirements for product candidate approvals;
•differing jurisdictions could present different issues for securing, maintaining or obtaining freedom to operate in such jurisdictions;
•potentially reduced protection for intellectual property rights;
•difficulties in compliance with different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations;
•changes in non-U.S. regulations and customs, tariffs and trade barriers;
•changes in non-U.S. currency exchange rates of the SEK, GBP, JPY, CNY, SGD and EUR and currency controls;
•changes in a specific country’s or region’s political or economic environment, including the implications of the United Kingdom’s withdrawal from the European Union, recent developments in China and the effects of recent increases in inflation and interest rates worldwide;
•trade protection measures, import or export licensing requirements or other restrictive actions by governments;
•differing reimbursement regimes and price controls in certain international markets;
•negative consequences from changes in tax laws;
•compliance with tax, employment, immigration and labor laws for employees living or traveling abroad, including, for example, the variable tax treatment in different jurisdictions of share options granted under a current or future equity incentive plan;
•workforce uncertainty in countries where labor unrest is more common than in the United States;
•difficulties associated with staffing and managing international operations, including differing labor relations;
•an outbreak of a contagious disease, such as coronavirus, which may cause us or our distributors, third party vendors and manufacturers and/or customers to temporarily suspend our or their respective operations in the affected city or country;
/36
•production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
•business interruptions resulting from geo-political actions, including war, such as the ongoing military conflict between Russia and Ukraine and between Israel and Hamas, and terrorism, or natural disasters including earthquakes, typhoons, floods and fires.
The United Kingdom’s withdrawal from the European Union may have a negative effect on global economic conditions, financial markets and our business, which could reduce the price of our common shares and ADSs.
Following the result of a referendum in 2016, the United Kingdom left the European Union on January 31, 2020, commonly referred to as “Brexit.” Pursuant to the formal withdrawal arrangements agreed between the United Kingdom and the European Union, the United Kingdom was subject to a transition period until December 31, 2021 (Transition Period), during which European Union rules continued to apply, while the future relationship between the United Kingdom and European Union was formally negotiated. The United Kingdom and the European Union have signed a EU-UK Trade and Cooperation Agreement, which became provisionally applicable on January 1, 2021 and became formally applicable on May 1, 2021 upon ratification by both the United Kingdom and the European Union. This agreement provides details on how some aspects of the UK and EU’s relationship will operate going forward; however there are still many uncertainties. The long- term effects of Brexit will depend in part on how the EU-UK Trade and Cooperation Agreement, and any future agreements signed by the United Kingdom and the European Union, take effect in practice. Such a withdrawal from the European Union is unprecedented, and it is unclear how the restrictions on the United Kingdom’s access to the European single market for goods, capital, services and labor within the European Union and the wider commercial, legal and regulatory environment, could impact our current and future operations and clinical activities in the United Kingdom.
Since we have a subsidiary in the United Kingdom, Olink Proteomics Limited, and employees located in the United Kingdom and a significant proportion of the regulatory framework in the United Kingdom applicable to our business and our products and services is derived from European Union directives and regulations, Brexit, now that the Transition Period is over, could materially impact the regulatory regime with respect to the development, manufacture, importation, approval and commercialization of our products and services in the United Kingdom or the European Union, as the United Kingdom legislation can now diverge from European Union legislation.
The uncertainty concerning the United Kingdom’s legal, political and economic relationship with the European Union following Brexit may also be a source of instability in the international markets, create significant currency fluctuations, and/or otherwise adversely affect trading agreements or similar cross- border co-operation arrangements (whether economic, tax, fiscal, legal, regulatory or otherwise).
If our laboratory facilities become damaged or inoperable or we are required to vacate or unable to access our existing facilities, our ability to conduct our laboratory processes and analysis and pursue our research and development efforts may be jeopardized.
We operate laboratory facilities located in Waltham, Massachusetts; Uppsala, Sweden; Umeå, Sweden; and through a third-party service provider in China. Our facilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including war, fire, earthquake, power loss, communications failure or terrorism, which may render it difficult or impossible for us to operate our platform for some period of time. The inability to perform our laboratory processes or to reduce the backlog that could develop if our facilities are inoperable, for even a short period of time, may result in the loss of customers or harm to our reputation, and we may be unable to regain those customers or repair our reputation in the future.
Furthermore, our facilities and the equipment we use to perform our research and development work could be unavailable inaccessible, or costly and time-consuming to repair or replace, which may increase backlog. It would be difficult, time-consuming and expensive to rebuild our facilities, to locate and qualify new facilities or license or transfer our proprietary technologies to a third party, particularly in light of licensure and accreditation requirements. Even in the unlikely event we are able to find a third party with such qualifications to enable us to conduct our laboratory processes, we may be unable to negotiate commercially reasonable terms.
We carry insurance for damage to our property and the disruption of our business, but this insurance may not cover all of the risks associated with damage or disruption to our business, may not provide coverage in amounts sufficient to cover our potential losses and may not continue to be available to us on acceptable terms, if at all.
/37
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because life sciences companies have experienced significant securities price volatility in recent years, with 2021 and portions of 2022 marking a period of extended share price volatility and decline. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business. Any such negative outcome could result in payments of substantial damages or fines, damage to our reputation or adverse changes to our business practices. Defending against litigation is costly and time- consuming, and could divert our management’s attention and our resources. Furthermore, during the course of litigation, there could be negative public announcements of the results of hearings, motions or other interim proceedings or developments, which could have a negative effect on the market price of the ADSs.
We identified material weaknesses in our internal control over financial reporting for our consolidated financial statements, and we may identify additional material weaknesses in the future that may cause us to fail to meet our reporting obligations or result in material misstatements of our financial statements. If we fail to remediate any material weaknesses or if we otherwise fail to establish and maintain effective internal control over financial reporting, our ability to accurately and timely report our financial results could be adversely affected.
The identified material weaknesses and remediation plans are further described in PART II, Item 15. CONTROLS AND PROCEDURES.
The process of designing and implementing an effective financial reporting system is a continuous effort that requires us to anticipate and react to changes in our business and the economic and regulatory environments and to expend significant resources to maintain a financial reporting system that is adequate to satisfy our reporting obligations. If we fail to develop or maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results, prevent fraud or meet our reporting obligations. As a result, investor confidence and the market price of our shares and our ADSs may be materially and adversely affected.
Risks Related to the Ownership of our Securities
Raising additional capital may cause dilution to holders of our common shares or ADSs, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We do not have any committed external source of funds or other support for our development efforts and we cannot be certain that additional funding will be available on acceptable terms, or at all. Until such time, if ever, as we can generate substantial product revenues, we expect to finance our operations through a combination of public or private equity offerings, debt financings, collaborations, strategic alliances, licensing arrangements and other marketing or distribution arrangements.
If we undertake financing arrangements in the future, the terms of any financing may adversely affect the holdings or the rights of holders of our common shares or ADSs and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of ADSs to decline. The sale or issuance of additional equity, convertible securities or warrants may dilute all of our existing shareholders and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a holder of ADSs. The incurrence of indebtedness could result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborators or others at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, financial condition and results of operations. Further, any additional fundraising efforts may divert our management from its day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our development programs or the commercialization of any of our product candidates, if approved, or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
Future sales, or the possibility of future sales, of a substantial number of the ADSs could adversely affect the price of the ADSs.
/38
ADSs representing the common shares issued and available for future issuance under our Amended and Restated 2021 Incentive Award Plan will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, the lock- up agreements and Rule 144 and Rule 701 under the Securities Act of 1933, as amended, or the Securities Act. If these additional ADSs are sold, or if it is perceived that they will be sold in the public market, the trading price of the ADSs could decline.
We expect that the price of the ADSs may fluctuate significantly and an active trading market for our common shares or ADSs may not be sustained.
The market price of the ADSs is likely to be volatile and could be subject to wide fluctuations in response to many risk factors listed in this section, and others beyond our control, including:
•actual or anticipated fluctuations in our financial condition and operating results;
•announcements by us, our partners or our competitors of new products, significant contracts, strategic partnerships, joint ventures, collaborations, commercial relationships or capital commitments;
•competition from existing products or new products that may emerge;
•failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public;
•issuance of new or updated research or reports by securities analysts or recommendations for our common shares;
•securities or industry analysts ceasing coverage of us, or publishing inaccurate or unfavorable research about our business;
•adverse regulatory announcements;
•disputes or other developments related to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies;
•commencement of, or our involvement in, litigation;
•fluctuations in the valuation of companies perceived by investors to be comparable to us;
•market conditions in our markets;
•manufacturing disputes or delays;
•any change to the composition of the board of directors or key personnel;
•expiration of contractual lock-up agreements with our executive officers and directors and shareholders;
•general economic conditions and slow or negative growth of our markets;
•the changing and volatile United States and global environments, including as a result of the COVID-19 pandemic and the public perception of pandemic associated risks;
•share price and volume fluctuations attributable to inconsistent trading volume levels of the ADSs;
•sales of the ADSs by members of our senior management and directors or our shareholders or the anticipation that such sales may occur in the future;
•securities or industry analysts ceasing coverage of us, or publishing inaccurate or unfavorable research about our business;
•investors’ general perception of us and our business;
•announcement or expectation of additional debt or equity financing efforts; and
•other factors described in this section of the Annual Report, many of which are beyond our control.
/39
These and other market and industry factors may cause the market price and demand for our ADSs to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their ADSs and may otherwise negatively affect the liquidity of the ADSs. Prior to our initial public offering of ADSs in March 2021, there was no public market for our ADSs and common shares. Even though our ADSs are listed on Nasdaq, there can be no assurance that an active trading market for ADSs will be sustained. In the absence of an active trading market for the ADSs, investors may not be able to sell their ADSs at or above the offering price or at the time that they would like to sell. The lack of an active trading market may also reduce the fair market value of the ADSs. In addition, the stock market in general, and life science companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies.
Holders of ADSs may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying common shares.
ADSs are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time when it deems expedient in connection with the performance of its duties. The depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or a governmental body, or under any provision of the deposit agreement, or for any other reason, subject to the right of ADS holders to cancel their ADSs and withdraw the underlying common shares. Temporary delays in the cancellation of your ADSs and withdrawal of the underlying common shares may arise because the depositary has closed its transfer books or we have closed our transfer books, and in other circumstances such as corporate actions including voting and dividend distributions. In addition, ADS holders may not be able to cancel their ADSs and withdraw the underlying common shares when they owe money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of common shares or other deposited securities. See "Item 12. Description of Securities Other Than Equity Securities - American Depositary Shares.”
Holders of the ADSs are not able to exercise the pre-emptive subscription rights related to the shares that they represent and may suffer dilution of their equity holding in the event of future issuances of our shares, convertible debentures or warrants.
Under the Swedish Companies Act, our shareholders benefit from a pre-emptive subscription right on the issuance of shares, convertible debentures or warrants for cash consideration only and not in the event of issuance of shares, convertible debentures or warrants against non-cash contribution or shares issued pursuant to convertible debentures or warrants previously issued by us. Shareholders’ pre-emptive subscription rights, in the event of issuances of shares against cash payment, may be disapplied by a resolution of the shareholders at a meeting of our shareholders and/or the shares may be issued on the basis of an authorization granted to the board of directors pursuant to which the board may disapply the shareholders’ pre-emptive subscription rights. Such shares may be issued at or above market value or below market value in the case of rights issues or pursuant to a resolution of the shareholders. The absence of pre-emptive rights for existing equity holders may cause dilution to such holders.
ADS holders would not be entitled, even if such rights accrued to our shareholders in any given instance, to receive such pre-emptive subscription rights related to the shares that they represent. Further, if we offer holders of our shares the option to receive dividends in either cash or shares, under the deposit agreement, ADS holders will not be permitted to elect to receive dividends in shares or cash but will receive whichever option we provide as a default to shareholders who fail to make such an election.
ADS holders do not have the same rights as our shareholders.
ADS holders do not have the same rights as our shareholders. For example, ADS holders may not attend shareholders’ meetings or directly exercise the voting rights attaching to the common shares underlying their ADSs. ADS holders may vote only by instructing the depositary to vote on their behalf. If we request the depositary to solicit your voting instructions (and we are not required to do so), the depositary will notify you of a shareholders’ meeting and send or make voting materials available to you. Those materials will describe the matters to be voted on and explain how ADS holders may instruct the depositary how to vote. For instructions to be valid, they must reach the depositary by a date set by the depositary. The depositary will try, as far as practical, subject to the laws of Sweden and the provisions of our articles of association or similar documents, to vote or to have its agents vote the deposited common shares as instructed by ADS holders. If we do not request the depositary to solicit your voting instructions, you can still send voting instructions, and, in that case, the depositary may try to vote as you instruct, but it is not required to do so. Except by instructing the depositary as described above, you will not be able to exercise voting rights unless you surrender your ADSs and withdraw the common shares. However, you may not know about the meeting enough in advance to withdraw the common shares. We cannot assure you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote your common shares. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that you may not be able to exercise voting rights and there may be nothing you can do if your common shares are not voted as you requested. In addition, ADS holders have no right to call a shareholders’ meeting.
/40
Holders of ADSs may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could result in less favorable outcomes to the plaintiffs in any such action.
The deposit agreement governing the ADSs representing our common shares provides that, to the fullest extent permitted by applicable law, ADSs holders waive the right to a jury trial of any claim they may have against us or the depositary arising out of or relating to our shares, the ADSs or the deposit agreement, including any claim under the United States federal securities laws. The waiver to right to a jury trial of the deposit agreement is not intended to be deemed a waiver by any owner or holder of ADSs of our or the depositary’s compliance with the United States federal securities laws and the rules and regulations promulgated thereunder.
If we or the depositary oppose a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable based on the facts and circumstances of that case in accordance with the applicable state and federal law. The enforceability of a contractual pre-dispute jury trial waiver in connection with claims arising under the federal securities laws has not been finally adjudicated by the United States Supreme Court. However, we believe that a contractual pre-dispute jury trial waiver provision is generally enforceable, including under the laws of the State of New York, which govern the deposit agreement. In determining whether to enforce a contractual pre-dispute jury trial waiver provision, courts will generally consider whether a party knowingly, intelligently and voluntarily waived the right to a jury trial. We believe that this is the case with respect to the deposit agreement and the ADSs. It is advisable that you consult legal counsel regarding the jury waiver provision before investing in the ADSs.
If you or any other owners or holders of ADSs bring a claim against us or the depositary in connection with matters arising under the deposit agreement or the ADSs, including claims under federal securities laws, you or such other owner or holder may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and/or the depositary. If a lawsuit is brought against us and/or the depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may result in a different outcome than a trial by jury would have had, including results that could be less favorable to the plaintiffs in any such action.
Nevertheless, if this jury trial waiver is not permitted by applicable law, an action could proceed under the terms of the deposit agreement with a jury trial. No condition, stipulation or provision of the deposit agreement or the ADSs serves as a waiver by any owner or holder of ADSs or by us or the depositary of compliance with any provision of the United States federal securities laws and the rules and regulations promulgated thereunder.
Because we do not anticipate paying any cash dividends on our common shares in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
We currently intend to retain all available funds and any future earnings to support operations and to finance the growth and development of our business, and do not anticipate paying any cash dividends on our common shares for the foreseeable future. In addition, the terms of any future debt agreements may preclude us from paying dividends. As a result, capital appreciation, if any, of our common shares or ADSs will be your sole source of gain for the foreseeable future. Furthermore, pursuant to Swedish law, the calculation of amounts available for distribution to shareholders, as dividends or otherwise, must be determined on the basis of our statutory accounts prepared in accordance with Swedish accounting rules. If the price of the ADSs or the common shares declines before we pay dividends, you will incur a loss on your investment, without the likelihood that this loss will be offset in part or at all by potential future cash dividends.
/41
Because we are a “controlled company” within the meaning of Nasdaq listing standards, our shareholders may not have certain governance protections that are available to shareholders of companies that are not controlled companies, which could make the ADSs less attractive to some investors.
Under Nasdaq rules, a company in which more than 50% of the voting power for the election of directors of the company is held by an individual, a group or another company will qualify as a “controlled company”. As of December 31, 2023, Knilo InvestCo AS, which is owned by several funds controlled by Summa Equity AB, owned directly or indirectly 77,284,718 of our common shares, which represents approximately 62% of our common shares outstanding. As a result, we are and will continue to be a “controlled company” under Nasdaq rules and will not be required to comply with certain Nasdaq rules that would otherwise require it to have: (i) a board of directors comprised of a majority of independent directors; (ii) compensation of its executive officers determined by a majority of the independent directors or a remuneration committee comprised solely of independent directors; and (iii) director nominees selected, or recommended for the board’s selection, either by a majority of the independent directors or a nominating committee comprised solely of independent directors.
We have not and do not expect to take advantage of the applicable exemptions under the Nasdaq corporate governance standards except to the extent we are exempt from such standards as a foreign private issuer; however, there can be no assurance we will not do so in the future if we are eligible. As such, our shareholders do not have and in the future will not have the same protections afforded to shareholders of companies that are subject to all of the corporate governance requirements under Nasdaq rules without regard to the exemptions available for “controlled companies.” Our status as a controlled company could make the ADSs less attractive to some investors.
Knilo InvestCo AS may have its interest in us diluted due to future equity issuances or its own actions in selling common shares, in each case, which could result in a loss of the “controlled company” exemption under Nasdaq rules. We would then be required to comply with those provisions of Nasdaq rules, subject to our election to comply with home country governance practices, as discussed below.
We qualify as a foreign private issuer and, as a result, we will not be subject to United States proxy rules and will be subject to reporting obligations under the Exchange Act, that, to some extent, permit less detailed and frequent reporting than that of a United States domestic public company.
We report under the Exchange Act as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to United States domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act, (ii) the sections of the Exchange Act requiring insiders to file public reports of their share ownership and trading activities and liability for insiders who profit from trades made in a short period of time and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form 20-F until 120 days after the end of each fiscal year, while United States domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation FD, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
As a foreign private issuer and as permitted by the listing requirements of Nasdaq, we rely on certain home country governance practices rather than the corporate governance requirements of Nasdaq.
We are entitled to rely on a provision in Nasdaq’s corporate governance rules that allows us to follow Swedish law regarding certain aspects of corporate governance. This allows us to follow certain corporate governance practices that differ in significant respects from the corporate governance requirements applicable to United States companies listed on Nasdaq. For example, we are exempt from Nasdaq regulations applicable to United States-listed companies regarding, and follow home country practice with respect to, the minimum quorum requirement for a meeting of shareholders, the requirement that non-management directors meet on a regular basis without management present, the requirement that the remuneration committee consist of independent members and the requirement that nominees of the Board are selected or recommended by a majority of the Board’s independent directors or by a nominations committee comprised of independent directors.
In accordance with our Nasdaq listing, our audit committee is required to comply with the provisions of Section 301 of the Sarbanes-Oxley Act, and Rule 10A-3 of the Exchange Act. Because we are a foreign private issuer, however, our audit committee is not subject to additional Nasdaq requirements applicable to listed United States companies, including an
/42
affirmative determination that all members of the audit committee are “independent” under the Nasdaq definition of independence. Furthermore, Nasdaq’s corporate governance rules require listed United States companies to, among other things, seek shareholder approval for the implementation of certain equity compensation plans and issuances of common shares, which we are not required to follow as a foreign private issuer. Therefore, our shareholders may be afforded less protection than they otherwise would have under corporate governance listing standards applicable to United States domestic issuers.
We may in the future lose our foreign private issuer status which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
We are a foreign private issuer and therefore we are not required to comply with all periodic disclosure and current reporting requirements of the Exchange Act applicable to United States domestic issuers. In order to maintain our current status as a foreign private issuer, either (a) a majority of our common shares must be either directly or indirectly owned of record by non-residents of the United States or (b)(i) a majority of our executive officers or directors may not be United States citizens or residents, (ii) more than 50% of our assets cannot be located in the United States and (iii) our business must be administered principally outside the United States. If we lose foreign private issuer status, we would be required to comply with the Exchange Act reporting and other requirements applicable to United States domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules.
The regulatory and compliance costs to us under United States securities laws if we are required to comply with the reporting requirements applicable to a United States domestic issuer may be significantly higher than the costs we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time-consuming and costly. We also expect that if we were required to comply with the rules and regulations applicable to United States domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our management team.
If we were to be classified as a passive foreign investment company, there could be adverse United States tax consequences to certain U.S. holders.
Under the U.S. Internal Revenue Code of 1986, as amended, we will be a “passive foreign investment company” for United States federal income tax purposes, or a PFIC, for any taxable year in which (1) 75% or more of our gross income consists of passive income or (2) 50% or more of the average quarterly value of our assets consists of assets that produce, or are held for the production of, passive income. If we are a PFIC for any taxable year during which a U.S. Holder (as defined below in "Item 10. Additional Information - E. Taxation - Material U.S. Federal U.S. Federal Income Tax Considerations for U.S. Holders”) holds our common shares or ADSs, the U.S. Holder may be subject to adverse tax consequences regardless of whether we continue to qualify as a PFIC, including ineligibility for any preferred tax rates on capital gains or on actual or deemed dividends, interest charges on certain taxes treated as deferred and additional reporting requirements.
A separate determination must be made after the close of each taxable year as to whether we are a PFIC for that year. Our status as a PFIC depends on the value of our assets and the composition of our income and assets. The total value of our assets for purposes of the asset test generally will be calculated using the market price of the ADSs, which may fluctuate considerably. Fluctuations in the market price of the ADSs may result in our being a PFIC for any taxable year. Our income for a taxable year will be affected by whether we receive certain milestone payments in such year, and whether certain gains from foreign currency exchanges are treated as qualifying income for purposes of the PFIC income test. Based upon the value of our assets and the composition of our income and assets, we do not believe we were a PFIC for the taxable year ended December 31, 2023 and, based on the current and expected composition of our income and assets and the value of our assets, we do not expect to be a PFIC for our current taxable year. However, no assurances regarding our PFIC status can be provided for the current taxable year or any past or future taxable years. For further discussion of the U.S. federal income tax consequences in the event we are classified as a PFIC, see "Item 10. Additional Information - Taxation - Material U.S. Federal Income Tax Considerations for U.S. Holders - PFIC Rules.”
The rights of our shareholders may differ from the rights typically offered to shareholders of a United States domestic corporation.
Under Swedish corporate law, except in certain limited circumstances, which require that a proposal for special review of accounts or a review of a specific item/topic as defined by shareholders requesting such review has been supported by shareholders representing not less than 10% of all shares in the company or one-third of the shares present at a shareholders' meeting, our shareholders may not ask for an inspection of our corporate records, while under Delaware
/43
corporate law any shareholder, irrespective of the size of such shareholder’s shareholdings, may do so. Shareholders of a Swedish limited company are also unable to initiate a derivative action, a remedy typically available to shareholders of United States domestic companies, in order to enforce a right of our company, in case we fail to enforce such right ourselves, other than in certain cases of board member/management liability under limited circumstances. In addition, a majority of our shareholders may release a member of our board of directors or our chief executive officer from any claim of liability we may have, including if such board member or our chief executive officer has acted in bad faith or has breached his or her duty of loyalty. However, a shareholder may bring a derivative action on behalf of our company against, among other persons, a member of our board of directors or our chief executive officer, provided that the circumstances of the act or omission giving rise to the claim of liability were not known to the shareholders at the time of such shareholder resolution, or if shareholders representing at least 10% of shares represented at the relevant shareholders' meeting have opposed such shareholder resolution. In contrast, most United States federal and state laws prohibit a company or its shareholders from releasing a board member from liability altogether if such board member has acted in bad faith or has breached such board member’s duty of loyalty to our company. Additionally, distribution of dividends from Swedish companies to foreign companies and individuals can be subject to non-refundable withholding tax, and not all receiving countries allow for deduction. Also, the rights as a creditor may not be as strong under Swedish insolvency law as under United States law or other insolvency law, and consequently creditors may recover less in the event our company is subject to insolvency compared to a similar case including a United States debtor. Finally, Swedish corporate law may not provide appraisal rights in the case of a business combination equivalent to those generally afforded a shareholder of a United States company under applicable United States laws.
For additional information on these and other aspects of Swedish corporate law and our articles of association, see ITEM
10. ADDITIONAL INFORMATION - B. Memorandum and Articles of Association. As a result of these differences between
Swedish corporate law and our articles of association, on the one hand, and United States federal and state laws, on the other hand, in certain instances, you could receive less protection as an equity holder of our company than you would as a shareholder of a United States company.
We are a Swedish company with limited liability. The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of United States jurisdictions.
We are a Swedish company with limited liability. Our corporate affairs are governed by our articles of association and by the laws governing companies incorporated in Sweden. The rights of shareholders and the responsibilities of members of our board of directors may be different from the rights and obligations of shareholders and members of boards of directors in companies governed by the laws of United States jurisdictions. In the performance of its duties, our board is required by Swedish law to consider the interests of our company, its shareholders, its employees and other stakeholders, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, the interests of our shareholders. See “Item 10. Additional Information - Memorandum and Articles of Association - Differences in Corporate Law”.
Claims of United States civil liabilities may not be enforceable against us.
We are incorporated under Swedish law. Certain members of our board of directors and senior management are non- residents of the United States, and a substantial portion of our assets and the assets of such persons are located outside the United States. As a result, it may not be possible to serve process on such persons or us in the United States or to enforce judgments obtained in United States courts against them or us based on civil liability provisions of the securities laws of the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce judgments obtained in United States courts against them or us, including judgments predicated upon the civil liability provisions of the United States federal securities laws.
The United States and Sweden do not currently have a treaty providing for recognition and enforcement of judgments (other than arbitration awards) in civil and commercial matters. Consequently, a final judgment for payment given by a court in the United States, whether or not predicated solely upon United States securities laws, would not automatically be recognized or enforceable in Sweden. In addition, uncertainty exists as to whether the courts in Sweden would entertain original actions brought in Sweden against us or our directors or senior management predicated upon the securities laws of the United States or any state in the United States. Any final and conclusive monetary judgment for a definite sum obtained against us in United States courts would not be automatically recognized. Instead, new proceedings would need to be initiated before the competent court in Sweden. However, a judgment obtained in the United States may still have a strong evidentiary weight in the Swedish proceedings, depending on the circumstances and the assessment of the court. If a Swedish court gives judgment for the sum payable under a United States judgment, the Swedish judgment will be enforceable by methods generally available for this purpose. These methods generally permit the Swedish Enforcement Authority (Sw. Kronofogden) discretion to prescribe the manner of enforcement. As a result, United States investors may not be able to enforce against us or certain of our directors any judgments obtained in United States courts in civil and commercial matters, including judgments under the United States federal securities laws.
/44
Our articles of association designate specific courts in the United States as the exclusive forum for certain United States litigation that may be initiated by our shareholders, which could limit our shareholders’ ability to obtain a favorable judicial forum for disputes with us.
Our articles of association provide that, unless we consent in writing to the selection of an alternative forum and without any infringement on Swedish forum provisions and without applying Chapter 7, Section 54 of the Swedish Companies Act (2005:551), the United States District Court for the Southern District of New York shall be the sole and exclusive forum for resolving any complaint filed in the United States asserting a cause of action arising under the Securities Act (Federal Forum Provision). In addition, our articles of association provide that any person or entity purchasing or otherwise acquiring any interest in our shares of capital stock will be deemed to have notice of and consented to the Federal Forum Provision; provided, however, that our shareholders cannot and will not be deemed to have waived our compliance with the U.S. federal securities laws and the rules and regulations thereunder.
We recognize that the Federal Forum Provision may impose additional litigation costs on shareholders in pursuing any such claims, particularly if the shareholders do not reside in or near the State of Delaware. Additionally, the Federal Forum Provision may limit our shareholders’ ability to bring a claim in a United States judicial forum that they find favorable for disputes with us or our directors, officers or employees, which may discourage the filing of lawsuits against us and our directors, officers and employees, even though an action, if successful, might benefit our shareholders. In addition, while the Delaware Supreme Court ruled in March 2020 that federal forum selection provisions purporting to require claims under the Securities Act be brought in federal court are “facially valid” under Delaware law, there is uncertainty as to whether other United States or Swedish courts will enforce our Federal Forum Provision. If the Federal Forum Provision is found to be unenforceable, we may incur additional costs associated with resolving such matters. The Federal Forum Provision may also impose additional litigation costs on shareholders who assert that the provision is not enforceable or invalid. The United States District Court for the Southern District of New York may also reach different judgments or results than would other courts, including courts where a shareholder considering a United States-based action may be located or would otherwise choose to bring the action, and such judgments may be more or less favorable to us than our shareholders.
General Risk Factors
Our employees, independent contractors, vendors and consultants may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk that our employees, independent contractors, vendors and consultants may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violate the regulations of the FDA, EMA and comparable foreign regulatory authorities, including those laws requiring the reporting of true, complete and accurate information to such authorities. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. We have adopted a Code of Conduct applicable to all of our employees, but it is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. Additionally, we are subject to the risk that a person could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, imprisonment, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non- compliance with these laws, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business, financial condition and results of operations.
We or our third parties upon whom we depend may be adversely affected by natural or man-made disasters or other business interruptions, and our business continuity and disaster recovery plans, or those of our collaborators, may not adequately protect us from the effects of a serious disaster.
Natural and man-made disasters and other events beyond our control, including events directly or indirectly resulting from climate change, could severely disrupt our operations, or those of third parties upon whom we depend, and have a material adverse impact on our business, results of operations, financial condition and prospects. If a natural disaster, power outage, or other event occurred that prevented us from using all or a significant portion of our headquarters, damaged critical infrastructure, such as our laboratory facilities or those of our collaborators, limited our or our collaborators’ ability to access or
/45
use our respective digital information systems or that otherwise disrupted our respective operations, it may be difficult or, in certain cases, impossible for us or our collaborators to continue our respective businesses for a substantial period of time. The disaster recovery and business continuity plans we and our collaborators currently have in place are limited and are unlikely to prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our respective disaster recovery and business continuity plans, which could have a material adverse impact on our business.
We have incurred and will continue to incur increased costs as a result of operating as a United States-listed public company, and our board of directors is required to devote substantial time to new compliance initiatives and corporate governance practices.
The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of Nasdaq and other applicable securities rules and regulations impose various requirements on non-U.S. reporting public companies, including the establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our board of directors and other personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations have and will continue to increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect that these rules and regulations may make it more difficult and more expensive for us to maintain or obtain director and officer liability insurance, which in turn could make it more difficult for us to attract and retain qualified members of our board of directors.
However, these rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Pursuant to Section 404 of the Sarbanes-Oxley Act, we will be required to furnish a report by our board of directors on our internal control over financial reporting. However, since we no longer qualify as an emerging growth company as of 31 December 2023, we are to comply with Section 404 of the Sarbanes-Oxley Act, we continue to be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and continue to implement a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude that our internal control over financial reporting is effective as required by Section 404 of the Sarbanes-Oxley Act and that material weaknesses may occur as described above.
/46
ITEM 4. INFORMATION ON THE COMPANY
A.History and Development of the Company
We were founded as a private limited company under the laws of Sweden on December 13, 2018 under the name Goldcup 18086 AB and registered with the Swedish Companies Registration Office on January 4, 2019. Knilo HoldCo AB’s operations (including subsidiaries; together the Companies or the Group) include development, production, marketing and sales of biotechnological products and services and related operations. Knilo HoldCo AB was incorporated on January 4, 2019. The Group was formed on March 7, 2019 when Knilo HoldCo AB acquired Olink OldCo AB (f/k/a Olink Proteomics Holding AB) through the subsidiary Olink Finance AB (the Olink Acquisition). The legal status of Knilo HoldCo AB was changed under Swedish law from a private limited company to a public limited company and the name was changed to Olink Holding AB (publ), which was registered with the Swedish Companies Registration Office on January 27, 2021.
On October 17, 2023, Thermo Fisher Scientific Inc. (NYSE: TMO) (“Thermo Fisher”) and Olink Holding AB (publ) (the Company) announced that their respective boards of directors have approved Thermo Fisher’s proposal to acquire the Company for $26.00 per common share in cash, representing $26.00 per American Depositary Share (ADS) in cash. On October 31, 2023, Thermo Fisher commenced a tender offer to acquire all of the outstanding common shares of the Company and all of the American Depositary Shares. The transaction, which is expected to be completed by mid-2024, is subject to customary closing conditions, including receipt of applicable regulatory approvals, and completion of the tender offer. As part of the transaction, Summa Equity AB, the Company's largest shareholder and additional shareholders and management, in aggregate holding more than 62% of the Company’s common shares, entered into support agreements agreeing to tender into the tender offer. Thermo Fisher expects to fund the acquisition using cash on hand and debt financing. Upon completion, the Company will cease being traded on Nasdaq, will become part of Thermo Fisher’s Life Sciences Solutions segment and will cease being a public reporting company.
We have twelve wholly owned subsidiaries - Olink Finance AB (f/k/a Knilo BidCo AB; Goldcup 18087 AB), a private limited company formed under the laws of Sweden in 2018, Olink OldCo AB (f/k/a Olink Proteomics Holding AB), a private limited company formed under the laws of Sweden in 2016, Olink Proteomics AB, a private limited company formed under the laws of Sweden in 2015, Agrisera Aktiebolag, a private limited company formed under the laws of Sweden in 1985, Olink KK, a company formed under the laws of Japan in 2019, Olink Biotech (Shanghai) Co., Ltd, a company formed under the laws of China in 2020, Olink Proteomics Inc., a Delaware corporation founded in 2015, Olink Proteomics Limited, a private company limited by shares formed under the laws of England and Wales in 2015, Olink Proteomics B.V., a private company formed under the laws of the Netherlands in 2016, Olink Proteomics GmbH, a limited liability company formed under the laws of Germany in 2018, Olink Proteomics SAS a private company formed under the laws of France in 2022 and Olink Proteomics SG Pte. Ltd a private company formed under the laws of Singapore in 2023.
Our registered office is located at Salagatan 16F, SE-753 30, Uppsala, Sweden, and our telephone number is +46
(0) 18 - 444 39 70. Our website address is www.olink.com. We have included our website address in this Annual Report solely as an inactive textual reference. The information contained on or accessible through our website is not incorporated by reference into this Annual Report. Additionally, the SEC maintains a website at www.sec.gov that contains reports and other information regarding registrants that make electronic filings with the SEC using its EDGAR system. Our filings made with the SEC are available on the SEC’s website.
B.Business Overview
Our purpose is to enable and accelerate the field of proteomics by providing a platform of products and services, that are deployed across major biopharmaceutical companies and leading clinical and academic institutions, to deepen the understanding of real-time human biology and drive 21st century healthcare through actionable and impactful science. Since our inception, we have served a customer base of more than 1,059 customer accounts in over 40 countries worldwide. We support three-quarters of the world’s 50 largest pharmaceutical companies (based on 2022 R&D spend), and many leading academic institutions. Many of these customers have carefully vetted and validated our technology before adopting Olink as part of their drug development programs. Our platform has been significantly validated, as evidenced by use of our products in studies that have been published in more than 1,600 peer-reviewed publications. We support our customers in understanding real-time human biology through proteomics by providing clarity on mechanistic biology and pathways that drive disease; by identifying novel and causal drug targets, which guides candidate drug development; by revealing predictive biomarkers for drug response, disease risk and outcomes, which identifies which patients have the potential to benefit the most from new therapies and treatments; and by detecting and characterizing indicators of disease and health to manage patient wellness more proactively. Our products and services play a role in decoding the biology of almost all disease areas and are used most frequently in immunology, oncology, neurology, cardiovascular and metabolic diseases.
Our current offerings are based on our proprietary and patented Proximity Extension Assay (PEA) technology, which enables researchers to use one platform from discovery to clinical applications utilizing a significant, established infrastructure of labs and installed instrumentation. PEA comprises three product lines: Explore, Target, and Focus,
/47
including our Signature instrument for gPCR read-out, each of which allows scientists to detect and quantify protein biomarker targets. Our library of protein biomarker targets is focused on circulating proteins with clinical utility, and we believe that it is among the world’s largest extensively validated protein libraries. To achieve a consistently high assay performance that does not compromise data quality of each protein biomarker target in our protein library, we have developed our own comprehensive validation framework with regulatory processes in mind, covering relevant, critical performance criteria such as specificity, sensitivity, dynamic range and precision. Our scalable high-throughput platform is differentiated from that of our competitors, as it is well-suited for a broad range of studies, from small to large scale, offering validated single-plex performance in a high-multiplex assay, designed to provide consistently high-quality data and address our customers’ needs across a broad range of applications. Hence, we believe the PEA platform is well positioned to support customers in the emerging high- throughput, high-plex proteomics use-cases and our customers utilize our platform for a variety of needs, from protein biomarker discovery in high-multiplex to clinical decision making. The first diagnostic protein signature for monitoring disease progression in Multiple Sclerosis (MS) based on PEA is being made available by Octave Bioscience in the diagnostics market. Test access is being offered as a service through their Clinical Laboratory Improvement Amendments (CLIA) certified lab based on custom developed kit products delivered by Olink. While our revenues and growth have historically been driven by the research market, we expect diagnostic applications of our platform will drive significant long- term growth.
According to a Nature publication from 2015, only approximately 20% of patients responded well to the top 10 highest grossing prescription drugs, with as many as 80% of patients experiencing non-responsiveness to the drugs’ intended benefits. Further, only 13.8% of compounds used in clinical trials make it through the drug development process to market. One factor that contributes to this low efficacy is that drugs may inadvertently target a confounding factor due to clinicians’ insufficient understanding of the pathophysiology driving the disease. As a result, drug developers and/or clinicians fail to identify a truly causal biological process and the drug target responsible for causing the disease. Furthermore, clinicians often classify disease too broadly, overlooking sub-populations of patients with different disease endotypes that require different treatment.
Twenty-first century healthcare, precision medicine, or personalized medicine, is an emerging practice of medicine that uses an individual’s molecular phenotype profile to guide and inform diagnostic decisions and to improve prediction of disease outcome and risk, leading to better informed decisions regarding disease prevention and therapeutic interventions for each individual, with the goal to provide the right treatment to the right patient at the right time. Precision medicine has the potential to enable clinicians to predict the most appropriate course of action quickly, efficiently, and accurately for individual patients, leading to improved outcomes for individual patients, as well as reduced costs and risks with shorter time to market for new drugs.
Over the past decade, genomics has been at the forefront of 21st century healthcare. While progress has been made in the field of genomics, there is a large unmet need to add additional insights into the molecular phenotype, particularly with respect to the proteome and proteins, which are the direct drivers of all biological processes in the human body and dynamic, real-time differentiators between health and disease, including dynamics affected by lifestyle and environment. Because proteomics is vastly more complex than genomics, researchers rely on sophisticated technologies to deliver actionable insights to advance the field. Unfortunately, existing legacy technologies have a number of limitations, including lack of specificity, especially in high-multiplex assays, lack of sensitivity and precision; limited dynamic range (which is the ability to reliably and simultaneously measure a wide range of concentrations); high sample consumption requirement; lack of scalability; low throughput; data complexity; and high cost. We believe that PEA has overcome these challenges, both from a technical perspective and cost perspective, and has the potential to move proteomics into a new paradigm.
Circulating protein biomarkers in blood represent an easily accessible sample type that both the biopharmaceutical industry and healthcare systems use. There are well known biomarkers used in diagnostics today, such as C-reactive protein (CRP) and Prostate-specific antigen (PSA), that are clinically actionable in that they mirror the biological processes of inflammation or malignancies, respectively. However, the number of clinically established biomarkers remains small while at the same time our appreciation of the complexity of diseases is increasing. Traditional disease classifications are increasingly being challenged and different sub-groups of disease endotypes that require different treatment strategies are continuously identified as diseases are being more molecularly defined. Hence, we believe this means that the need for new circulating biomarkers has never been greater and will require the ability to sample the dynamic plasma proteome in sufficient depth, breadth and specificity since most likely patterns or signatures of multiple proteins will be required to properly reflect the complexity of disease.
As illustrated by Figure 1 below, the plasma proteome contains high-abundant “classical plasma proteins” as well as tissue leakage and low-abundant proteins such as interleukins and cytokines. Although proteins at all abundance levels provide valuable information, we believe that PEA’s ability to provide granular insights into the many low-abundant circulating proteins will allow scientists to better identify novel and causal drug targets guiding candidate drug development. PEA has the potential to reveal predictive biomarkers for drug response, disease risk and outcomes, which may enable scientists to identify which patients have the potential to benefit the most from new therapies and treatments, and aid scientists in detecting and characterizing indicators of disease and health so that they can more proactively manage patient wellness. We believe that 21st century healthcare will be driven by clinically actionable, low-abundant circulating proteins mirroring biological processes in the human body and PEA will play an important role in that process.
/48
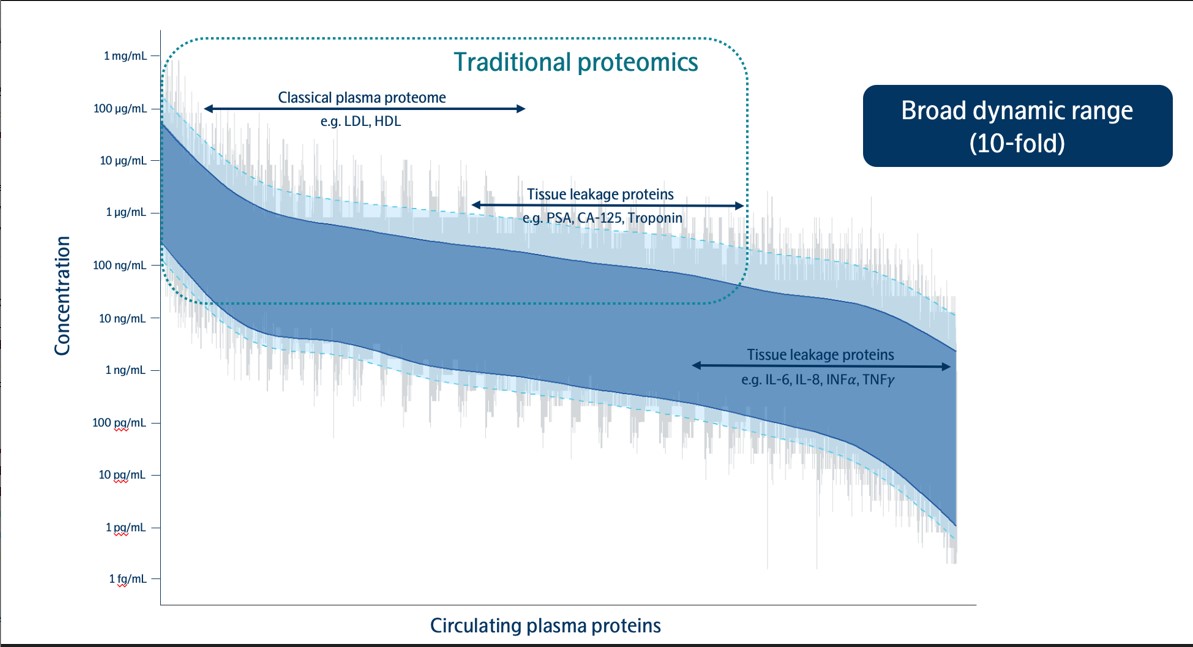
Figure 1. Illustration of Olink’s library of protein biomarker targets covering a wide dynamic concentration range (y-axis) and including proteins (x-axis) measured in mg/ml to pg/ml. The highlighted proteins are examples of select PEA success stories in identifying important biomarkers and in which concentration they typically occur.
PEA has enabled the interrogation of low-abundant circulating proteins in high throughput and high-multiplex with high data quality, which enables scientists to discover novel and subtle individual differences in the plasma proteome. With these insights enabled by PEA, our customers are making revolutionary findings that we believe change our understanding and definitions of diseases. We believe that this research was enabled by PEA and would not have been possible five years ago.
We believe our proprietary and patented PEA technology has broad application in proteomics at large scale in high-multiplex discovery as well as in more targeted clinical trial and diagnostic applications. Compared to many other technologies, PEA can enable faster, better-informed decisions in human protein biomarker research by providing protein biomarker targets in high-multiplex with an assay performance that does not compromise on data quality. To achieve a consistently high assay performance for all biomarker targets in our library, our proprietary and comprehensive validation framework, which was developed with regulatory processes in mind, includes critical performance criteria such as specificity, sensitivity, dynamic range, scalability, lack of interference, reproducibility and precision. Most of our assay products require only 1 µL or less of sample volume, which is approximately 20 to 1,000 times less than the sample volume required by certain other proteomics technologies. This sample volume efficiency combined with our high-multiplexing capabilities is designed to provide high throughput at a reasonable cost, which is important for any platform used in large-scale proteomics where researchers are looking to analyze thousands of proteins in thousands of samples in the same study over weeks or months. Our customers have validated the utility and value of our technology and products, as evidenced by use of our products in studies that have been published in more than 1,100 peer-reviewed publications and by expanding usage of our products in clinical trials. Most importantly, our technology provides our customers with one platform they can use from protein biomarker discovery in high-multiplex to clinical decision making and diagnostics, with broad applicability across substantially all relevant biological sample types.
Our technology today incorporates a constantly growing library of highly validated protein biomarker targets of which our customers currently can access approximately 5,400 to detect and quantify in their samples. Our current library focuses on proteins detectable in plasma in order to provide clinically relevant, actionable and meaningful insights to our customers. 200 of our assays is incorporated into the Flex product library, 1,100 into the Target product line and approximately 5,400 is incorporated into the Explore product line. We plan to grow the library as far as commercially and scientifically relevant over time. Accordingly, we believe that as we grow our library in size and depth, we would be able to provide a holistic and high-resolution view of the plasma proteome encompassing the most relevant biological processes and pathways in the human body. In fact, when overlaying our library with the Reactome, a comprehensive database of biological pathways, our 5,400 protein biomarker targets, available in Explore, offer a complete coverage of all major pathways, such as the immune system or metabolism. We also believe that our PEA technology’s ability to provide this holistic, broad and deep, real-time view of human biology with high data quality and throughput will allow us to further differentiate ourselves from established and emerging high-multiplex proteomics technologies. Based on our platform’s broad capabilities we expect to continue increasing the size our antibody library over time, and also plan to include protein biomarker targets in our library that are not typically detectable in plasma. Our library expansion process includes consultations with KOLs and our customers and a rigorous curation process undertaken by our data scientists, who apply machine learning methods to identify and select the most biologically impactful and clinically relevant biomarkers.
/49
We believe we are the only company providing a holistic proteomic offering from broad protein biomarker discovery in high- multiplex through clinical decision making and diagnostics. We offer kit products in three products lines. Our Explore line with next generation sequencing (NGS) readout offers a fully automated process utilizing our complete library for large- scale studies with market-leading throughput. The Explore offering has the potential to enable researchers to complete the multi-omics perspective, by combining genomics, transcriptomics and proteomics, on the same underlying technology platform. Our Target line with quantitative polymerase chain reaction (qPCR) readout is optimized for targeted research and clinical development at a smaller scale using relative or absolute quantification. Our Focus offering of custom- developed kit products allows customers to define their protein profile of interest for clinical applications such as clinical trials or diagnostic products.
For customers that prefer outsourced proteomics analysis, we also offer Analysis Service, which includes assay execution and bioinformatics. Our experts support customers with study design, assay preparation, sample analysis, data processing, and we provide a comprehensive report with quality-controlled results. In order to best serve our global customers in the most timely and efficient manner possible, we operate Analysis Service labs out of our Waltham, Massachusetts and Uppsala, Sweden locations and through a third-party service provider in China.
We estimate that our addressable market is $42 billion. This market can be broadly classified into research and diagnostics based on the applications of our products and the types of customers we serve. Currently, the main driver of demand for our products and services is the research community’s unmet need for methods to better facilitate prediction of drug response and disease risk and outcomes. We are able to support customers throughout their entire journey from discovery to clinical decision making on one technology platform and believe that we are well positioned to become the protein enabler of multi-omics, especially on NGS. The Total Addressable Market (TAM) estimates were developed by us with support from third party market research and management consulting firms.
•Research. We estimate the research opportunity, our core market today, is $23 billion and define this opportunity as the addressable protein biomarker discovery research spend by biopharmaceutical companies and academia, consisting of a high-plex segment and low and mid-plex segment. The high-plex segment is expected to evolve through large-scale screening projects, including the emerging field of population proteomics where researchers build on the genomics research from the past decade by adding proteins. The research opportunity is defined as the estimated technology spend in the life science tools market for genomics and proteomics technologies that we can address with our existing and anticipated products. Each technology segment (such as multiplex immunoassays, mass spectrometry or NGS) has been segmented based on region, customer segment and use- case (i.e. the purpose for using the technology) before determining the share of spend addressable by us. In June 2020, we launched Olink Explore as a service through our Analysis Service labs utilizing NGS readout for PEA. Starting in early 2021, we made Explore available as NGS-based kit products to existing and new customers who are end-users of the estimated installed base, now estimated to more than 8,900 addressable Illumina systems. NGS is a technology platform that we expect will continue its high-growth trajectory, and we estimate that the installed base of addressable Illumina systems will grow to more than 10,000 by 2027, driven by Illumina's continued innovations and new competition entering the field, which are expected to drive down the cost of sequencing, and new NGS applications such as PEA. We believe that multi-omics will be an important growth driver of the NGS-market as a whole and our ability to enable multi-omics including proteins on NGS will represent an especially attractive growth opportunity for us. The latest addition to the Explore product line is Explore HT launched mid 2023, with a substantially simplified workflow offering 4* higher throughput than it´s predecessor. The low- and mid-plex segment consists of more targeted protein biomarker discovery research extending through all phases of clinical studies. In 2022, we continued to improve our product offering and platform. In Q4 2022 we launched the Olink Flex offering, to complement our Target products in our mid-plex product portfolio. Olink Flex offers our customers the possibility to mix and match up 21 proteins, out of a library of approximately 200 validated assays focusing on inflammatory protein biomarker targets, in absolute quantification. In 2023, we added Target 48 Mouse Cytokine to our mid-plex offering to serve translational studies. The low sample volume required (1 ul) is unique on the market and is enabled through the PEA technology. We estimate that the number of addressable proteomics labs will exceed 6,000 by 2027. The ability to leverage existing instrumentation and infrastructure removes significant barriers to customer adoption, which we believe will translate into more rapid market penetration.
•Diagnostics. We estimate the diagnostics opportunity is $19 billion and define this market as selected, relevant diagnostic applications for in vitro diagnostics (IVD) and laboratory developed tests (LDT). The diagnostics opportunity is defined as the end-market value of the clinical diagnostics biomarker markets, including LDTs, that we can address with our existing or anticipated products. The market was segmented by the biomarkers or methodologies applied in diagnostics by disease area (such as cardiovascular diseases or laboratory immunoassays) before determining the share of spend addressable by us. Our goal is to enable biopharmaceutical companies and IVD and LDT providers by providing access to high-quality multiplexed proteomics diagnostics products that can be applied in diagnostic settings. We estimate that there are approximately 43,000 hospitals in the OECD countries which we believe would benefit from such novel
/50
diagnostics solutions in the future. The first diagnostic protein signature for monitoring and disease progression in Multiple Sclerosis (MS) based on PEA is being made available by Octave Bioscience.
•We have a successful history of developing molecular technologies based on commercializing pioneering academic research. We were founded in 2016, and in March 2019 we were acquired by Summa Equity AB, a Nordic private equity firm, which enabled the next step in our development. Since inception, more than 1,059 customer accounts in over 40 countries have utilized our products and services. A customer account is defined as one company (which is the case for the majority of our industry customers) or a department at a larger institution (which is often the case for larger universities where multiple customer accounts can exist). Further, since inception we have supported three-quarters of the world’s largest 50 pharmaceutical companies (valued by R&D spend), including 19 of the largest 20 and many leading academic institutions. We consider the majority of more than 1,000 customer accounts to be reoccurring customers, as they buy in regular intervals, even if not annually, and as an example, revenues from our customers obtained in 2016 represent approximately 23% of our revenues for the year ended December 31, 2023 and have grown at an average annual growth rate of 31% as of December 31, 2023 and 31% as of December 31, 2022. From 2020 to 2022, the average revenues from new customers represented approximately 18% share of total revenues. As of December 31, 2023, we had 707 employees, including a recently increased commercial team of 245 individuals and an R&D team of 82 individuals. The majority of our employees operate out of our Uppsala, Sweden headquarters. We also have secondary headquarters in Waltham, Massachusetts and a growing footprint across Singapore, China and Japan.
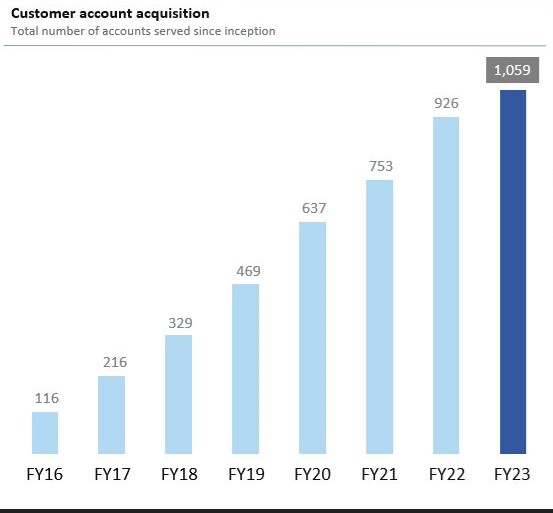
Figure 2. Evolution of Olink’s customer accounts served since inception.
Our customer-focused science and operational models have translated into robust performance, including growing our revenues to $169.6 million, 21.3% growth compared to the 2022 fiscal year; incurring a net loss of $31.6 million; and generating an adjusted EBITDA loss of $5.8 million for the year ended December 31, 2023. During 2022 and 2023, we
/51
increased our investment in human capital, which resulted in 166 new employees (net) in 2022 and 125 new employees (net) in 2023. We expect to continue to accelerate investment in human capital over the coming years.
Adjusted EBITDA is a measure not calculated in accordance with IFRS® Accounting Standards (IFRS). For more information regarding our use of adjusted EBITDA and reconciliations of adjusted EBITDA to operating loss, the most directly comparable financial measure calculated in accordance with IFRS Accounting standards, see "Item 5. Operating and Financial Review and Prospects - Non-IFRS Reconciliations " herein.
Our Competitive Strengths
Our historical and anticipated future growth are underpinned by a set of competitive strengths that we believe will not only allow us to accelerate the field of proteomics, but also to increasingly establish ourselves as the leading player in the emerging proteomics space. Our competitive strengths include:
•Our proprietary PEA technology enables industry leading assay performance in high- multiplex and high- throughput proteomics. Progress in proteomics has historically been hampered by the lack of technologies that can provide reliable and consistent assay performance in high-multiplex. Our proprietary methods of combining affinity-based detection of proteins with optimized methods for amplification and detection of nucleic acids is the reason why PEA can overcome these challenges. Our PEA technology succeeds where other technologies have failed as it enables high-multiplex, high-throughput and cost-efficient proteomics without compromising data quality. We believe PEA is the only technology combining high performance for each protein biomarker target across specificity, sensitivity, dynamic range, scalability, precision and interfering factors, all in high-multiplex, resulting in highly reproducible and actionable data. We believe this gives us a technological advantage in proteomics and a differentiation in the market that we will continue to build on in the future.
•We have an extensively validated and rapidly growing library of high-quality actionable protein biomarker targets. To date, we have developed a library of protein biomarker targets, of which 5,400+ are available in our Explore product, that we selected with input from KOLs and customers as well as published proteomics research. We focused initially on the most actionable and clinically relevant proteins accessible in the human plasma, which are thought to be associated with major disease areas. Our targets include low-abundant inflammation proteins, actively secreted proteins, organ-specific proteins leaked into circulation, drug targets (established and from ongoing clinical trials) and proteins detected in blood by mass spectrometry. Our platform incorporates robust analytical validation data that we publish on our website in an open-access format. We drive growth and optimization of our library through our internal antibody development capabilities. Our goal is to continue to invest heavily in scaling our library and we plan to increase the number of highly validated protein biomarker targets to grow as far as commercially and scientifically relevant over time.
•By design, our platform supports a customer from protein biomarker discovery research to diagnostic applications, all on one single underlying technology platform. Our platform is well-suited for small-to-large- scale protein biomarker studies, offering solutions for relevant applications from the largest screening projects to highly targeted, hypothesis-driven studies. Depending on the customer’s needs, we can offer validated single-plex performance in high multiplex for consistently high data quality regardless of the use-case. For large-scale and high-plex studies, we use the NGS readout, which provides an ideal solution for customers who wish to run high- throughput studies with large numbers of human serum or plasma samples against our complete library of proteins. For more targeted research and clinical applications, we use the qPCR readout, which provides a high-quality and flexible offering using one or several panels most relevant to the subject of study. Our flexibility and scalability allow us to offer our customers one technology platform through all phases of drug development and research, and across a wide range of biological sample types, with built-in consistency and reproducibility.
•We have long-standing and close-knit relationships with our significant and growing customer base and leading KOLs across relevant disease and applications areas. We have cultivated close-knit relationships that we believe are based on trust with our customers, as we have developed our products and solutions for, and in collaboration with, our customers. From leading research universities to top biopharmaceutical companies, our customers have rigorously vetted and validated our technology, and we believe the reliability and high quality of our offering has driven high customer engagement and loyalty. Many of the most prominent KOLs in proteomics are our supporters and promoters, as evidenced by use of our products in studies that have been published in more than 1,100 peer-reviewed publications and by expanding use in our customers’ clinical trials. Combined with the quality of our technology offering, our team of talented professionals provides world-class service and support, and are fully committed to helping our customers succeed.
•Our next-generation proteomics product, Explore, integrates with existing NGS workflows enabling accelerated adoption of the platform. We emphasize flexibility and usability across our platforms in order to drive accessibility and broad adoption. Explore, uses Illumina’s sequencing technology as a readout platform and has an installed base estimated to more than 8,900 systems to generate proteomic data. Whilst our technology is most commonly used on Illumina's systems today, we are agnostic in terms of NGS platform. By combining PEA
/52
with NGS, we hope to become the scaled proteomics enabler of multi-omic signatures that builds on genomics work from the past decade, while providing the research and clinical community with a seamless multi-omics solution to predict disease outcomes and drug response.
•Our purpose-built readout platform, Olink Signature Q100, has the potential to make PEA more accessible to customers through thousands of existing proteomics labs. We initially began using an existing qPCR readout platform provided by Standard Biotools (previously Fluidigm) for our Target and Focus products, both internally and in the many external labs we work with. To accelerate the adoption of this part of our portfolio, we developed Olink Signature Q100, a purpose-built qPCR readout instrument optimized for PEA, which was made commercially available in June of 2021. We believe that Olink Signature Q100 will drive an accelerated market adoption of PEA among the more than 4,800 addressable proteomics labs and adoption of our Target, Focus and Flex product lines.
•Our robust proteomic analysis software and evolving open-access cloud-platform, Olink Insight, has the potential to further establish our position enabling a community driven understanding of real-time human biology by accelerating proteomics. Our deep experience in protein biomarker discovery combined with our team of analytics experts and software developers allow us to provide our customers with proprietary self-service software and analytical tools for data analysis and comparison with robust quality control. We believe that the ease of analyzing, and interpreting PEA data is an important point of differentiation, especially in larger population proteomics studies. Additional software processing capabilities include the identification and verification of individual protein profiles, which reveal real-time biology status of the patient. We designed Olink Insight to work with Olink data, offering a range of data visualization options that are precise, easy to interpret, and provide an excellent overview of complex data sets, all to accelerate the time it takes for scientists' to generate actionable insights from their data. The reliability and ease of our analytical solutions enable the efficient assessment of data quality and rapid identification of potential issues. Olink Insight allows our customers to openly share and contribute data and insights to the research community to collectively accelerate the field of proteomics.
Our Growth Strategy
Our strategy centers on driving the market adoption of PEA by lowering barriers to adoption and actively engaging with our community of KOLs and customers to accelerate proteomics. Our growth strategy includes:
•Accelerate market adoption and scale our footprint to establish market leadership in the field of proteomics by making PEA more widely accessible worldwide. As more researchers come to experience the benefits of PEA, we see an opportunity to bring PEA closer to the customer and establish our platform in new labs while expanding the Olink ecosystem. As we continue to grow, we plan to scale our kits business as we believe this offering will enable us to significantly broaden access to our proteomic solutions. We will work to continue to expand our customer base, both within our current markets and in new use-cases, applications, and fields, as well as in new geographic markets.
•Aggressively grow our library of validated, high-quality and actionable protein biomarker targets and optimize our content. While our initial library has focused on what we believe to be the most clinically relevant and actionable proteins to maximize the impact we have on the field of proteomics and in 21st century healthcare, our goal is to develop a library that grow as far as commercially and scientifically relevant biomarker targets. We plan to continue developing the most relevant content based on biological interest and high-likelihood of clinical applicability in major disease areas, in conjunction with KOLs, and applying machine learning methods to the selection process. We are leveraging our in-house antibody development and increasingly utilizing recombinant antibodies and expanding their use in protein biomarker discovery. Our acquisition of Agrisera AB in 2020 has and will continue to allow us to rapidly increase the number of biomarker targets in our library through our own antibody development capabilities. In addition, we have included some commercially available antibodies from a number of select vendors to build out the library.
•Firmly establish Olink as the proteomics standard by building on, expanding and accelerating our well- established KOL relationships. Our technology was born out of work by leading scientists in protein research, and we strive to maintain that heritage as we innovate and bring new offerings to market. We plan to continue working with key thought leaders in proteomics to test new concepts, generate more proof points and bring about advancements. We see an opportunity in our KOL relationships to help define the future of proteomics and establish Olink as the proteomics standard.
•Expand and deepen the Olink eco-system by leveraging Olink Insight, our cloud platform, to develop a unique proteomics data source together with our research community. We are pushing transparency initiatives aimed at generating, open access datasets based on Olink through collaborations and make complex data such as proteogenomic data, available to the research community. Our goal is to accomplish this through our cloud platform, Olink Insight, creating the most accessible and comprehensive source of proteomics knowledge for the scientific community. We believe this initiative has the potential to solve current challenges within
/53
proteomics, by enabling the community to perform more efficient data analysis, generate results more quickly and reach actionable conclusions faster. We view our platform as a way to bring our customers, the broader scientific community and Olink closer together in an eco-system where we can accelerate proteomics together.
•Expand our product portfolio to make our offering the broadest and most accessible in proteomics, addressing unmet needs in the research community. We plan to invest heavily to maintain our edge as a technology leader in the proteomics field with an offering that can address our customers’ unmet needs. We are continuing to develop PEA to increase its applicability across platforms, configurations, and use-cases. We listen intently to feedback from our customers, and we aim to optimize workflows for a seamless customer experience.
•Capture the diagnostics opportunity by supporting our customers’ journeys from discovery to clinical decision making. Collectively, our Explore, Target, Flex and Focus offerings cover all stages of research. With our reputation for excellence in protein discovery research firmly established, we see significant opportunity to build our presence in clinical development and clinical decision making. The purpose of our Focus offering is to enable our customers to develop customized kit products for protein signatures based on PEA and improve clinical decision making. Over time, we could directly participate in discovery and clinical decision making by collaborating and partnering in the clinical end-markets, and in some instances, by investing and developing our own products for proprietary clinical applications.
•Scale up the Olink organization for the future. We believe that our strong purpose-driven culture and talented team of professionals are key pillars to our success. From January 1, 2023, through December 31, 2023, 125 new (net) employees joined Olink. We intend to continue to accelerate investment through 2024 and over the coming years, including investing heavily in our infrastructure and continue to grow employee headcount to support our growth opportunity and strategy, while maintaining industry-leading employee satisfaction. We plan to continue investing in the development of our employees and promoting our culture of customer service and support through innovation, quality, rigor and transparency, as well as fostering our shared vision to enable understanding of real-time human biology.
•Accelerate our reach and rate of adoption through new business models, partnerships and by deepening successful customer relationships. We regularly reevaluate Olink’s role in the proteomics value chain in order to apply the most appropriate business and commercial models to advance our market position. We believe we have the ability and expertise to enter into strategic partnerships and acquisitions across the proteomics value spectrum, and our product offering is easily adaptable to a variety of commercial models and scientific collaborations that allow us to scale our efforts and accelerate proteomics research. We regularly look for opportunities to engage in strategic partnerships with leading global companies to continue expanding Olink’s role in advancing proteomics.
Our Technology
We believe our proprietary and patented PEA technology has the characteristics necessary for broad application in proteomics at large scale in discovery and in more targeted ways in clinical trials and diagnostic applications. Compared to many other technologies, PEA can enable faster, better-informed decisions in research by enabling detection and quantification of protein biomarkers in high-multiplex and high-throughput with an assay performance that does not compromise on data quality.
Our Market Opportunity
We estimate that our addressable market is $42 billion, and this market can be broadly classified into research and diagnostics categories based on the applications of our products and the types of customers we serve. We estimate the research opportunity, our core market today, is $23 billion and define this opportunity as the addressable protein biomarker discovery research spend by biopharmaceutical companies and academia, consisting of a high-plex segment and low and mid-plex segment. We estimate the diagnostics opportunity is $19 billion, consisting of selected, relevant diagnostic applications for IVD and LDT. The Total Addressable Market (TAM) estimates were developed by us in connection with support from a third party market research and management consulting firm, and additional market research acquired from a third party market research firm.
Currently, the main driver of demand for our products and services is the research community’s unmet need for methods to better facilitate prediction of drug response and disease risk and outcomes. To address these needs, there will be a need to move beyond just genomics by adding proteins to develop multi-omics signatures. Our ultimate goal is to enable our customers to take protein signatures from discovery to clinical decision making in the current decade. We anticipate that the significant and growing investment required for this will come from both academia and biopharmaceutical companies, each currently representing 50% of research spend. In the future, to realize the potential for 21st century healthcare, we expect biopharmaceutical companies to direct a larger share of their research budgets towards proteomics and multi-omics applications. Accordingly, we expect biopharmaceutical companies to make up a larger market share in the future and drive a higher share of the market growth as they search for clinical multi-omics applications to enable the
/54
ability to predict drug responders and disease outcomes. With our ability to support customers throughout this entire journey on one technology platform, we believe we are in the best position to become the protein enabler of multi-omics in this market.
The Research Opportunity
We estimate the research opportunity is $23 billion, representing a significant growth opportunity for us as we believe we have just begun scratching the surface of our full potential. The research opportunity is defined as the estimated technology spend in the life science tools market for genomics and proteomics technologies that we can address with our existing and anticipated products. Each technology segment (such as multiplex immunoassays, mass spectrometry or NGS) has been segmented based on region, customer segment and use-case (i.e. the purpose for using the technology) before determining the share of spend addressable by us. PEA is a relatively young technology that we believe we can grow by converting users of other proteomics technologies to PEA and increasingly participating in the genomics markets where proteomics can add additional insights and potentially provide a better scientific answer. We characterize the research opportunity in two segments: high-plex and low- and mid-plex. High-plex refers to the high-throughput and large scale proteomics use- cases where customers are analyzing up to many thousands of proteins in up to many thousands of samples in the same studies. Low- and mid-plex refers to more targeted research. For example, in mid-plex, customers are typically analyzing hundreds to thousands of proteins in up to many thousands of samples, such as in clinical trials. In low-plex, customers have typically identified a number of proteins of interest, often referred to as a protein signature, of five to ten proteins that they would like to focus on.
We expect the high-plex segment to evolve through large-scale screening projects, including the emerging field of population proteomics where researchers build on genomics research from the past decade by adding protein data at large scale. Technological innovation, and recently with new competitors in the market, has considerably reduced the cost of gene sequencing, accelerating its use and driving an increase in the identification of possible genetic targets and biomarkers for earlier and more precise disease diagnosis, patient selection, treatment and monitoring. Since our inception, we have observed a consistent trend towards higher plex, the research community still severely lack knowledge and importance of most circulating proteins in the transition from health to disease. As we deliver higher plex at a lower cost per data point and with “clinical” quality, we have expanded our market by adding more content for the research community to explore. We expect to continue building on this trend and, since early 2021, we have started to externalize Explore through NGS-based kit products to existing and new customers who are end-users of the estimated installed base estimated to more than 8,900 addressable Illumina systems. NGS is a technology platform that we expect will continue its high-growth trajectory, and we estimate that the installed base of addressable Illumina systems will grow to more than 10,000 by 2027, driven by Illumina's continued innovations and new competition entering the field, which are expected to drive down the cost of sequencing, and new NGS applications such as PEA. We believe that multi-omics will be an important growth driver of the NGS market as a whole and our ability to enable multi-omics with proteins on multiple NGS platforms will represent an especially attractive growth opportunity for us. In addition, we believe our ability to access this existing infrastructure and participate in the rapidly growing NGS landscape will contribute to the accelerated adoption of our products.
The low- and mid-plex segment consists of more targeted protein biomarker discovery research, where one studies around 10-100 proteins, extending through all phases of clinical studies. This is where we initially built our business, and in 2021 we took the next step towards making our Target and Focus products more accessible to approximately 4,800 addressable proteomics labs with the order start of our purpose-built qPCR-based detection system for PEA, Olink Signature Q100. The instrument was launched in June 2021 and began shipping to customers during the fall of 2021. We estimate that the number of addressable proteomics labs will grow to approximately 5,800 by 2027. Even in the low and mid-plex segment, we expect the trend towards higher plex to continue in this market segment, driving an increase in focused research that will, on average, result in a higher number of protein biomarker targets being studied, which we believe plays into the benefits of PEA. The unmet needs of this market center on improving specificity and increasing sensitivity with lower sample consumption in higher plex. We believe that our new qPCR system will allow us to effectively target existing proteomics labs.
The Diagnostics Opportunity
We estimate the diagnostics opportunity at $23 billion, consisting of selected, relevant diagnostic applications for IVD and LDT. The diagnostics opportunity is defined as the end-market value of the diagnostics biomarker markets, including LDTs, that we can address with our existing or anticipated products. The market was segmented by the biomarkers or methodologies applied in diagnostics by disease area (such as cardiovascular diseases or laboratory immunoassays) before determining the share of spend addressable by us. Our goal is to enable biopharmaceutical companies and IVD and LDT providers by providing access to high-quality multiplexed proteomics diagnostics products that can be applied in diagnostic settings. The first diagnostic protein signature for monitoring and disease progression in Multiple sclerosis (MS) based on PEA is being made available by Octave Bioscience in the diagnostics market. Test access is being offered as a service through their Clinical Laboratory Improvement Amendments (CLIA) certified lab based on custom developed kit products delivered by Olink. The end-market pricing is expected to be determined by reimbursement, such as from insurance companies. We believe that PEA can play a meaningful role in clinical decision making in five major disease
/55
areas: immunology, oncology, neurology, cardiovascular and metabolic diseases. We also believe PEA can be valuable in markets where proteins already play a role in the product offering and can also be highly relevant to current solutions for genetic testing and other application areas. We anticipate that we will increasingly participate in this market by enabling our customers to transition to clinical decision making with PEA and by collaborating with customers to develop and commercialize proprietary clinical applications.
Our Products and Services
Our PEA technology is available to customers through products: Explore, Target, Flex and Focus, enabling the detection and quantification of thousands of protein biomarker targets in different configurations, with different workflows depending on the type of research conducted. Figure 3 below is an overview of the current product portfolio of available products and comparison of key differences. The products are available as kit products or as a service through our Analysis Service labs.
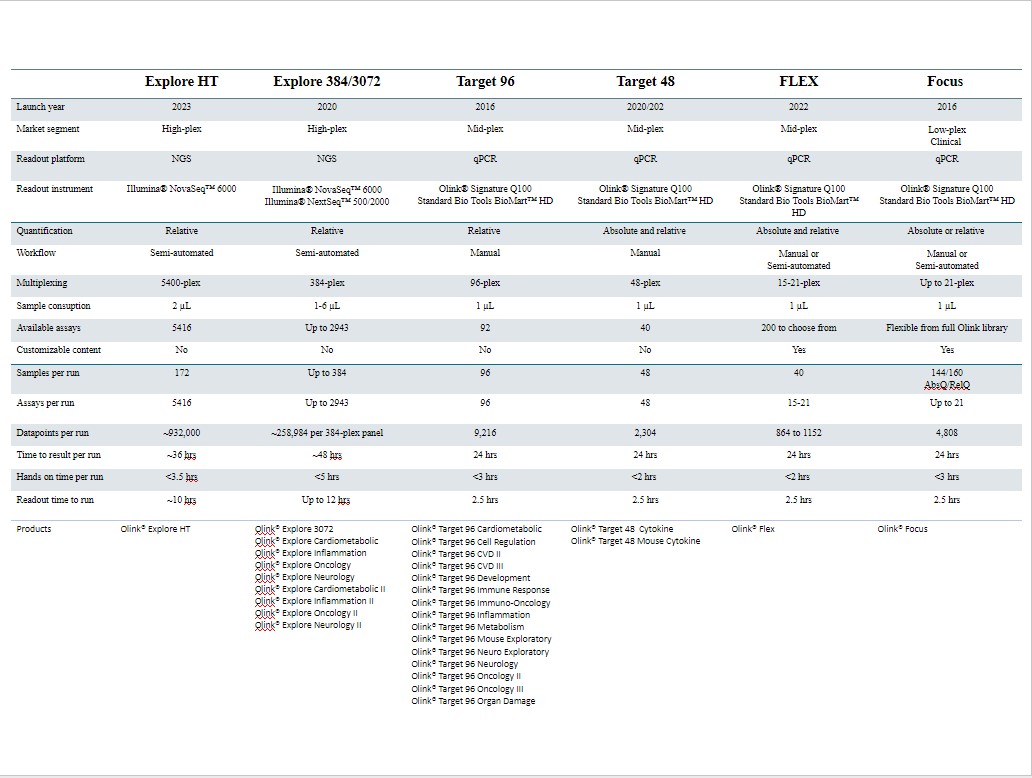
Figure 3. Olink portfolio of products at a glance with relevant specifications.
The latest addition to Explore offering, Explore HT, was launched mid 2023. Explore HT includes 5,400 biomarker targets. A new NPX Explore software was launched at the same time with a completely updated quality control module and with an additional tool, the CLI interface, to support automated workflows at our customers. Target 96, Target 48 and Flex kit products have a similar composition, but are slightly different as they are smaller kits and are for qPCR workflow. Flex kits is our customized mid-plex solution where you mix and match you own panel of 15-21 analytes from a library of 200.
In Olink Insight, our open-access online portal that supports users in understanding and interpreting their proteomics data, we have added features such as data analytics tools and new data stories such as the Disease Atlas.. We designed Olink Insight to work with Olink data, offering a range of data visualization options that are precise, easy to interpret, and provide an excellent overview of complex data sets, all to accelerate the time it takes for scientists' to generate actionable insights from their data.
/56
We develop a Validation Data Package for each Olink product that we make available to both customers and general visitors to our website. The reports contain a detailed dataset showing the performance for each protein biomarker target in the product across each performance criteria in the validation framework. These reports provide transparency to customers, which we think is an important part of our value proposition, and further reinforce the trust we have developed. For the Target products the reports can be downloaded, while for the Explore products the reports, given their size and complexity, availability will be online only. Figure 4 below illustrates the contents of a typical Validation Data Package.

Figure 4. Overview of the Validation Data Packages developed for each Olink product.
Olink Explore
In June 2020, we launched Olink Explore as a service through our Analysis Service labs utilizing NGS readout for PEA. Since early 2021, Explore has been made available to customers worldwide as distributed kit products. The product line was developed for the high-plex market segment to meet our customers’ need for large scale proteomics with high- throughput and high-multiplex. Explore has received a strong reception since its launch and is the largest revenue stream of all products.
Explore HT was launched in July 2023. Our earlier products, including Explore 372 and Explore 384, were each designed to be particularly relevant for cardiovascular and metabolic diseases, oncology, neurology or inflammation, and remains an important part of our Explore offering with its modular design. Explore is run on Illumina´s NovaSeq systems or as individual 384 runs on Illumina´s NextSeq systems.
Olink Target
We launched our Olink Target product line at our inception in 2016, and it has been the pillar of our business to date. It initially utilized qPCR readout on Fluidigm’s Biomark HD system and, starting in 2021 we took the next step towards making our Target and Focus products more accessible with the commercialization of our purpose-built qPCR-based detection system for PEA, Olink Signature Q100. With Target we service the low- and mid-plex segment and address its need for more targeted discovery research at various levels of plex, often targeting certain specific disease areas. We have, therefore, designed each of our 15 Target 96 products to be particularly relevant to specific disease areas.
/57
Historically, a customer would run anywhere from one to 13 products in parallel to cover up to 1,161 protein biomarker targets per sample in one experiment.
In October 2020, we launched our first Target 48 Cytokine product with absolute quantification in 48-plex. Target 48 was specifically developed for careful monitoring of the immune system and downstream applications in clinical trials, where the understanding of protein concentrations at the individual level is more important than understanding the differences in protein concentrations for larger groups. In 2023, we launched Target 48 Mouse Cytokine, aimed at translational studies, with a competitive differentiator of a minute (1ul) sample volume requirement.
Olink Flex (part of Target)
Olink Flex targets low-plex applications and use-cases and was launched in November 2022. Customers who are interested in a select number of protein biomarker targets will benefit from a fully flexible made-to-order panel running on Olink Signature Q100. The offering allows customers to combine 15 to 21 human proteins using absolute quantification in one biomarker panel. Using an online panel builder in Olink Insight, customers can freely pick and choose 15 to 21 pre-validated assays from roughly 200 human protein biomarkers covering major biological pathways such as inflammation, immuno-oncology and oncology, neurology, and cardiovascular disease. The product format allows users to measure 40 samples simultaneously, with readout in pg/mL and Normalized Protein eXpression (NPX) while only requiring 1µl of sample. We believe that Olink Flex represents a significant innovation in the low-plex market that will make the PEA technology even more accessible for scientists and can serve as an entry level product for modern higher multi-plex proteomics.
Olink Focus
Our Olink Focus product line consists of custom developed solutions for customers that have identified a small number of proteins of interest, or a protein signature, to focus on. The customer can choose up to 21 protein biomarker targets from our full library and apply relative or absolute quantification, and we will then develop and validate the product for them. Focus is typically used for very targeted research, often late- stage clinical trials, and when the customer sees a path towards clinical applications.
We developed our first Focus product in 2017 with a protein signature used for patient stratification of women with different stages of ovarian cancer. The customer worked with Olink from early discovery through verification and validation of replication cohorts.
Olink Signature
In June 2021 we launched Olink Signature Q100, our own qPCR readout platform, with shipping beginning during the fall of 2021. The system is purpose built for PEA and we believe it will make our Target and Focus kit products more widely accessible in the market. As qPCR has proven to be a highly suitable platform for PEA, we believe we have incorporated the best of the technology. The Olink Signature Q100 is a cost efficient, ultra-light and nimble benchtop system with a modern design and equivalent or better performance properties than Fluidigm’s Biomark HD system. Olink Signature Q100 is the readout platform used for our Target and Focus product lines, both for external installations and in our Analysis Service labs.
Olink Analysis Service
We operate service labs out of Uppsala, Sweden, and Waltham, Massachusetts, and offer our services through a third- party service provider in China. We have highly skilled Analysis Service staff and data scientists who will support the customers in the entire process. Our typical turnaround time, from sample in to data out, is four to six weeks. The Analysis Service offering includes:
•Study design and consultation;
•Sample preparation and assay execution; and
•Data processing and QC.
Olink Data Science services
As a complement to our standard Analysis Service offering and to ensure we support all our customers from initial study design to biological conclusion, we offer advanced data analysis and bioinformatics services. Depending on customer needs, our data science team can support customers with customized statistical and data analysis. Our bioinformatics offering includes:
/58
•Access to a data science team specialized in working with NPX data;
•Customizable solutions to support customer needs;
•Fast analysis of data using state-of-the-art statistical methods; and
•Data analysis consultation and training..
Olink NPX data processing software
Olink offers several purpose-built software designed for customers who run Olink’s products in their own facilities and is required to quality control runs using Olink’s built in control and generate Olink’s proprietary NPX format.
NPX Signature
The NPX Signature software allow import of data directly from Olink’s Signature Q100 instrument without any prior processing or annotation needed by the user. This allows our customers to use one software to process data from the Olink Target, Focus and Flex products, which include both relative and absolute quantification measurements of protein concentration. The software allows the operator the import multiple runs, perform quality control and generate result reports within one tool with focus on ease of use and data quality. The process is outlined in Figure 5 below.
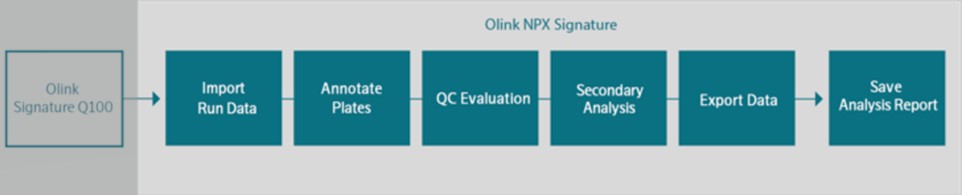
Figure 5: Overview of data processing from Signature Q100 run to generated analysis report.
NPX Explore[MSA1]
Olink’s NPX Explore software is designed to generate Olink’s proprietary NPX format and full analysis reports for Olink’s high plex Explore panels. The whole pipeline can be run on premise by the customer. Data processing is initiated with raw next generation sequencing data being prepared in a preprocessing step using Olink’s proprietary software bcl2counts. The resulting counts file is thereafter imported to NPX Explore. Multiple NGS runs and panels can be processed simultaneously, including quality control, normalization procedures and final export of NPX data and analysis reports. An overview of processing of Olink Explore data can be seen in Figure 6 below.

Figure 6: Overview of data processing from NGS readout of Olink Explore panels to final NPX data and analysis report exports.
/59
Data analysis tools for the Olink community
Olink provides tools to support downstream data analysis to our customers and community allowing faster time to result and biological insight when using Olink’s products and generated data. Olink’s data science team develops and maintains the open source R package Olink Analyze that is available in public repositories. This analysis package allows our customers to easily import data from all Olink products and perform data analysis including visualizations and statistical modelling. The package is a versatile toolbox that enables easy handling of all Olink data.
Olink Insight
Olink Insight is an innovative scientific that addresses the complex challenges of proteomic data. Built on scientific data and rich features, Olink Insight empowers users on their journey from experiment planning and design to post-run data analysis. Starting from a scientific question, Olink Insight will guide product recommendations and provide scalability of studies. Analytical and visualization tools, shared data sets such as Disease Atlas and Normal Ranges, combined with shared expertise and other data sources, further provides users with a faster time to biological conclusions and to understand disease.
Its significant features include:
•Pathway Explorer. This feature graphically represents the proteome and coverage of Olink panels in biological pathways. It explores which proteins are involved in a specific pathway to help with interpretation of data and generation of actionable results. It also reveals and helps users understand connections between proteins and human biology. Built on Reactome, an open-source peer reviewed database.
•Annotation. This feature helps users draw biological conclusions from proteomic data by listing biomarker-specific information about tissue specificity, drug target, how the corresponding protein functions at the molecular level, where in the cell it functions, what biological processes it helps to carry out, and variability in a normal cohort.
•Data Stories. The feature showcases Olink´s 'best practice analysis' on a real and relevant publicly available data set demonstrating how results can be analyzed and visualized in practice.
•Normal Ranges. Is the largest public data source that describes the natural variability of plasma protein levels across more than variability in more than 50,000 samples from three different cohorts and continents. This information allows the user to take more data driven decision when selecting biomarkers and designing studies as well as comparing results to this comprehensive resource.
•Disease Atlas. This feature allows users to compare and explore observed protein expression profiles in the most common diseases, generated by protein profiling of patients. The first version of the Disease Atlas focuses on 15 different cancer types, and will later expand to include other common diseases, such as cardiovascular, neurological, infectious, and autoimmune diseases; as well as other conditions.
•Design and analyze. Olink Insight provide tools to assist the user to easily design powerful studies and quickly analyze Olink data for common study design to reach result faster.
•Publication Explorer. This feature provides users with a shortlist of significant publications based on a selected biomarkers from a study, including scientific abstracts where significant "hits" may co-occur with each other, with key words, and diseases.
•Build Custom Panels. Allows users to build and requests quote for custom panels with Olink Flex or select pre-configured panels by Olink´s experts for important research areas such as inflammation, immune-oncology and aging.. Customers can pick and choose 15 to 21 pre-validated assays from roughly 200 human protein biomarkers covering major biological pathways such as inflammation, immuno-oncology and oncology, neurology, and cardiovascular disease.
Research and Development
We seek to improve our proprietary products and services to develop a broad and accessible proteomics product portfolio and intend to allocate an increasing level of investment to R&D over the coming years with a significantly broader scope
/60
than in past years. We are focused on lowering barriers for adoption across a number of detection platforms and improving our scalable offering for downstream clinical applications.
PEA’s unique capability of creating a DNA barcode representing the targeted protein biomarker in a sample allows for agnostic read-out across various qPCR and NGS platforms, as well as arrays. We evaluate and select which platforms to enable for amplification and detection of the DNA barcodes. To date, we have used the Biomark HD system from Standard BioTools. Olink's Signature Q100 system, the NovaSeq 6000 from Illumina and, most recently since early 2021, the NextSeq 550 and 2000 from Illumina, In addition, we are continuosly exploring new opportunities based on factors including use-case, application area, installed base, throughput and cost. Recently, multiple new companies have entered the NGS market and we have demonstrated the compatibility of these new sequencing chemistries with our Explore products, which will provide customers with the ability to pursue their platform of choice, We are also enabling more options on which liquid handlers to use. Enabling more detection platforms and sample preparation solutions is consistent with our platform agnosticity strategy. In terms of multiplex scalability, we currently offer products in 24, 48, 96 and 384 plex independently or in various combinations to cover a larger part of our library in the same study. We intend to continue to increase our multiplexing capabilities over time and we regularly evaluate market opportunities in the low-, mid- and high- plex markets and seek to develop products to target any market segment and unmet need. Applying our in-house developed and validated proprietary oligo framework and conjugation chemistry, we can rapidly and efficiently build new products in various multiplexing formats based on emerging market needs and amplification/detection opportunities.
We also focus on rapidly expanding our library of validated, high-quality protein assays driving growth in the biomarker discovery space. Our library growth is driven by several factors including input from key opinion leaders from important disease and application areas, customer feedback, and new publications from biomarker studies. To accelerate the pace of assay development and to increase the control over our supply chain, we acquired Agrisera in early 2020. We anticipate Agrisera will help us improve assay content and to continue expanding our assay library further.
Scientific Affairs
A key part of our strategy has been to work closely with scientific thought leaders to drive the focus and content of our library, product development, validation strategies and data analyses.
We see a strong trend in our market to collaborate and share data to enable the understanding of real-time human biology and accelerate the field. Based on that trend and the technological advances we have made, we have been selected to work with various consortia across our industry. Examples of these include:
•SCALLOP. The SCALLOP consortium is a collaborative framework across biopharmaceutical companies and academia for discovery and follow-up of genetic associations, with proteins exclusively measured on the Olink platform. Each SCALLOP member works on human study collections from the general population, clinical trials or patients with certain diseases such as coronary artery disease, rheumatoid arthritis, bipolar disease, heart failure, dementias, or metabolic syndrome. The aim of the SCALLOP consortium is to identify novel molecular connections and protein biomarkers that are causal in diseases to identify novel drug development targets (illustrated in Figure 7). To date, 35 Principal Investigators (PIs) from 28 research institutions have joined the effort, including more than 57 different cohorts, which now comprises a summary level data set on genetic variations to protein level associations for more than 70,000 patients or controls. PIs of studies using Olink proteomics and genome-wide genotyping data are eligible to participate in the consortium.
Figure 7. Overview of SCALLOP’s ambition
•The UKB Pharma Proteomics Project (UKB-PPP). The Olink Explore platform was used to measure circulating concentrations of thousands of proteins in approximately 54,000 individuals from the UK Biobank, one of the world’s largest genetic resources. This project is funded by a consortium of thirteen biopharmaceutical companies. By the end of the second half of 2022, Olink delivered approximately 54,000 samples to the UK Biobank, including bridging samples run on the Explore 3072 Platform. This is enabling the availability of millions of protein measurements in a matter of months, with the ultimate goals of enabling better understanding of disease processes and supporting
/61
innovative drug development. Initial results from the UK Biobank Pharma Proteomics Project were published online to the bioRxiv preprint server in June 2022. Combined with the vast collection of existing UKB data, this initiative offers the research community an open-access proteomics resource of unprecedented breadth and depth, enabling deep plasma proteomics analysis to accelerate development of novel biomarkers and therapeutics. Given the significant need of the scientific community to access large datasets for biomarker discovery and validation purposes, UKB-PPP is a unique opportunity supporting future high-impact studies and publications.In March 2023 the UKB released Olink Eplore assay data publicly available for use by the public research community. The data included normalized expression measures for 1,500 proteins for 54,000 participant samples. In October 2023 this was expanded to include more than 3,000 proteins measured on the Olink Explore 3K platform. Since then there has been at least 16 peer-reviewed studies published on the UKB proteomic data set.
The study also includes an effort on COVID-19 where approximately 1,500 longitudinal samples from participants who tested positive for COVID-19 and approximately 1,500 samples from participants who tested negative for COVID- 19 were included for analysis.
•Foundation of the National Institute of Health (FNIH). Olink has been selected as partner of the FNIH Biomarker consortium consisting of biopharmaceutical companies and academic researchers with the ultimate goal of identifying biomarkers for diagnosis, prognosis and progression of disease. To create and lead cross-sector efforts that validate and qualify biomarkers and other drug development tools to accelerate better decision making for the development of new therapeutics and health technologies.
•CORAL. The CORAL consortium is a collaborative framework aiming to accelerate the identification of proteins and mechanisms for neurological diseases, as well as the translation of novel biomarkers for neurological diseases to the clinic. Each CORAL member will be working on human study collections, based on Olink data, from the general population, clinical trials or patients and is focused on neurological diseases such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), epilepsy, and other diseases. Together members can validate their findings and identify leads for cross-disease markers and mechanisms. Currently, around 25 parties are part of CORAL representing academic researchers, biopharma companies and foundations working together to accelerate discovery, validation, and implementation of biomarkers for neurological conditions.
•COLLIBRI. The COLLIBRI consortium consists of biopharmaceutical companies with current or development-stage drugs for Inflammatory Bowel Disease (IBD), and prominent clinical researchers treating patients with IBD. By applying genomic and proteomic approaches, the goal of the consortium is to identify novel drug target candidates and biomarkers to predict drug response and disease outcome in order to improve drug development efforts and patient outcomes.
We also work in close concert with leading researchers across many fields to promote the importance and significance of high-quality, large-scale proteomics. Examples include:
•COVID. We conducted a study with Massachusetts General Hospital and the Broad Institute analyzing data on ~1,500 proteins from 384 participants, 306 of whom tested positive for COVID-19 and 78 of whom tested negative for COVID-19. We supported the discovery of a protein signature predictive of disease outcome and were able to facilitate the stratification of more severe patients (death or severely ill) at the time of entry to the emergency care unit. Further detail regarding this study is illustrated in Figure 8 below.
/62

Figure 8. Results of COVID-19 case study.
•Melanoma. We conducted a study with Massachusetts General Hospital in which we performed plasma proteomic analysis of over 700 proteins at three serial time points (day 0, six weeks and six months) on 174 metastatic melanoma patients treated with immune checkpoint blockade (ICB). We supported the identification of predictive protein biomarkers’ responses to ICB in these patients. Further detail regarding this study is illustrated in Figure 9 below.
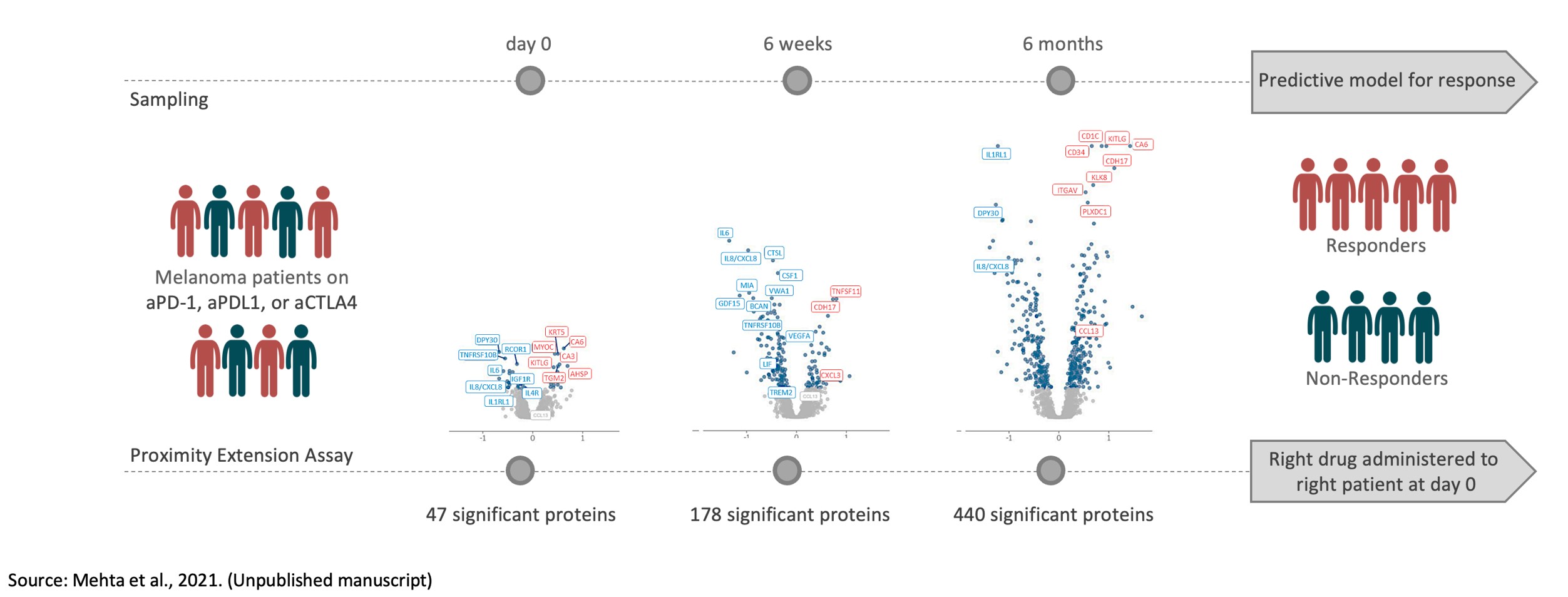
Figure 9. Results of immunotherapy case study.
•Ovarian cancer. Ovarian cancer is the eighth most common cancer among women and has a 5-year survival of only 30–50%. Current tests have insufficient sensitivity and specificity for early detection of ovarian cancer, so there is a strong need to identify biomarkers for early detection of ovarian cancer to increase survival. To address this urgent need, we supported the discovery of protein signature for highly accurate detection of early- and late-stage ovarian cancer with higher specificity and sensitivity compared to today’s diagnostic method (CA-125) and replicated in a second verification cohort. With these new findings, opportunities are opening up to establish screening programs, based on self-collected clinical samples, as a cost-efficient solution for early detection of ovarian cancer.
•Alzheimer disease (AD). Alzheimer’s disease (AD) is the most common age-related neurodegenerative dementia accounting for 60–80% of demented patients which makes it a threat to the aging population. Researchers at the Amsterdam University Medical Center measured ~700 proteins, using Olink’s Target 96 panels, to better understand proteomic changes in cerebrospinal fluid of patients followed by development of a custom Focus panel with an
/63
improved ability to distinguish Alzheimer disease patients from controls. With this result, it is possible to start to define the potential added value of these markers in routine diagnosis and clinical trials of drugs targeting Alzheimer disease.
•Inflammatory skin disease research. We have worked with leading KOLs on inflammatory skin diseases since our inception. Dermatological diseases such as psoriasis, eczema, and alopecia are of great medical and socioeconomic significance, and are contributing to the nonfatal disease burden in global health care. These diseases are often chronic and can have major physical and emotional impacts on sufferers, significantly reducing their quality of life. While such conditions may be classified as “skin diseases” their underlying pathophysiology is complex, involving systemic inflammation and autoimmune processes. Exemplifying this complexity, diseases such as psoriasis are thought to be associated with increased cardiovascular risk, including myocardial infarction and stroke. Consequently, dermatological conditions represent both a challenge when it comes to penetrating their underlying biology and developing new and better therapies, and also an opportunity to gain insights into a wider range of mechanistically related diseases. We believe protein biomarkers have the potential to play an important role in the field of inflammatory skin diseases and can contribute to these goals by improving our biological understanding and helping us to develop more effective, targeted treatments for patients in the future. Our PEA technology has successfully been applied in studies aiming to interrogate systemic inflammation of moderate and severe disease by evaluating skin and blood abnormalities, in children and in adults, and for monitoring efficacy, safety and pharmacokinetics of drugs for inflammatory skin diseases. By applying broad proteomics analysis using our PEA technology, researchers have also been able to characterize skin proteomic signatures and its relationship with the blood proteome and genome to increase the understanding of the pathology of these complex diseases.
•The wellness study. To achieve the goal of precision medicine, not only do different molecular profiles need to be understood in disease populations, but they must also be understood in the context of healthy populations. This especially applies to the stability of molecular profiles among healthy individuals over time, as this will clarify what qualifies as a “normal range” of clinical parameters in health and disease research. We supported a large Swedish initiative with leading KOLs at Karolinska Institute and Royal Institute of Technology on a large wellness study. Longitudinal analysis of blood profiles from healthy individuals helps us understand how they vary between individuals as well as within an individual over time. Comprehensive studies using our PEA technology on a longitudinal wellness cohort with healthy individuals have been conducted with analysis of blood molecular profiles based on proteomics, transcriptomics, lipidomics, metabolomics and autoantibodies. Results show high variation between individuals across different molecular readouts, while the intra-individual baseline variation is low. The analysis demonstrated that each individual had a unique and stable plasma protein profile throughout the study period and that many individuals also showed distinct profiles with regards to the other omics datasets, with strong underlying connections between the blood proteome and the clinical chemistry parameters. Results from proteogenomic studies also using our PEA technology have shown that many proteins detected in blood are determined at birth by genetics, which is important for efforts aimed at understanding the relationship between plasma proteome profiles and human biology and disease. In conclusion, the results support that health should be viewed at the level of the individual, rather than being more generalized. Moreover, the stability of the proteomics data emphasizes its potential to empower routine lab tests by providing more biologically relevant insights when interpreting data in both translational and clinical settings. Researchers conclude that the path forward lies in developing a comprehensive longitudinal molecular patient profile.
•Octave Bioscience. Octave Bioscience very carefully selected protein biomarkers for quantifying and monitoring disease activity of patients with Multiple Sclerosis (MS) through rigorous Feasibility, Discovery, Development and Validation stages, screening >1,400 proteins in more than 1,500 patients from multiple cohorts. For each of the final protein signature, the following parameters were characterized: accuracy, precision, robustness, sensitivity, MS reference ranges, interference, diurnal variability, cross-reactivity and stability of reagents and serum samples. The final analytically and clinically validated protein panel meets Octave’s stringent analytical performance specifications and measures the serum concentrations of 20 proteins associated with four biological pathways involved in MS pathophysiology. The custom panel built on Olink’s PEA platform allows rapid, accurate measurement of absolute protein concentrations in blood serum and have been demonstrated to reliably quantify and monitor disease activity of MS. Test access is being offered as a service through Octave's Clinical Laboratory Improvement Amendments (CLIA) certified lab. In October 2022 Octave´s precision care solution including the blood-based biomarker test was launched commercially and has since gained traction among MS clinics. In September 2023 Octave announced it will be conducting an observational study sponsored by Biogen. In November Octave received a $10 Million Grant from The Michael J. Fox Foundation to develop and validate a protein biomarker test for Parkinson´s disease.
Commercial
Olink was founded in 2016. Since our inception, we have served a customer base of more than 1,059 customer accounts in over 40 countries worldwide and we have supported three-quarters of the world’s largest 50 pharmaceutical companies by 2022 research and development spending, including 20 of the largest 20, and many of the most prestigious academic institutions, where many of these customers have carefully vetted and validated the technology before adopting Olink as part of their drug development programs. This vetting and validation process includes, for example, running Olink side-by-side with other proteomics technologies with samples that have been depleted for certain or all proteins, spike-ins of other proteins in certain concentrations, running samples in duplicates or triplicates, and then comparing results to evaluate
/64
which platform reports the highest quality data for the purposes of the research questions. The utility and actionability of our platform have been demonstrated by our strong and growing adoption by a community of researchers within academia, government, and the biopharmaceutical and biotechnology industries. Our customers primarily include academic, government, biopharmaceutical, biotechnology, service provider, and other institutions focused on life science research. We sell our products and services globally primarily through our own global direct sales force organized across our three market regions: Americas, EMEA and APAC. As of December 31, 2023, we had 707 employees of which the commercial team consisted of 245 individuals. The commercial team operates out of our Uppsala, Sweden headquarters and locally in other European markets such as the UK and France. We also have secondary headquarters in Waltham, Massachusetts and a growing footprint across Singapore, China and Japan. Expanding our commercial team and strengthening our sales and marketing capabilities are top priorities for us as a company and we expect to allocate significant investment to these parts of the organization in the next few years. We have taken significant steps forward in 2023, adding 37 as a net of new employees in the commercial team from January 1, 2023 through December 31, 2023, with respect to our capabilities, including investing heavily in our infrastructure and will continue to expand our organization as needed to support our growth potential and strategy. Figure 10 is an illustration of our commercial model and how it has evolved over time. We believe that the combined accomplishments of our commercial team since inception have positioned Olink for continued growth as we believe that they contribute to a positive feedback loop.
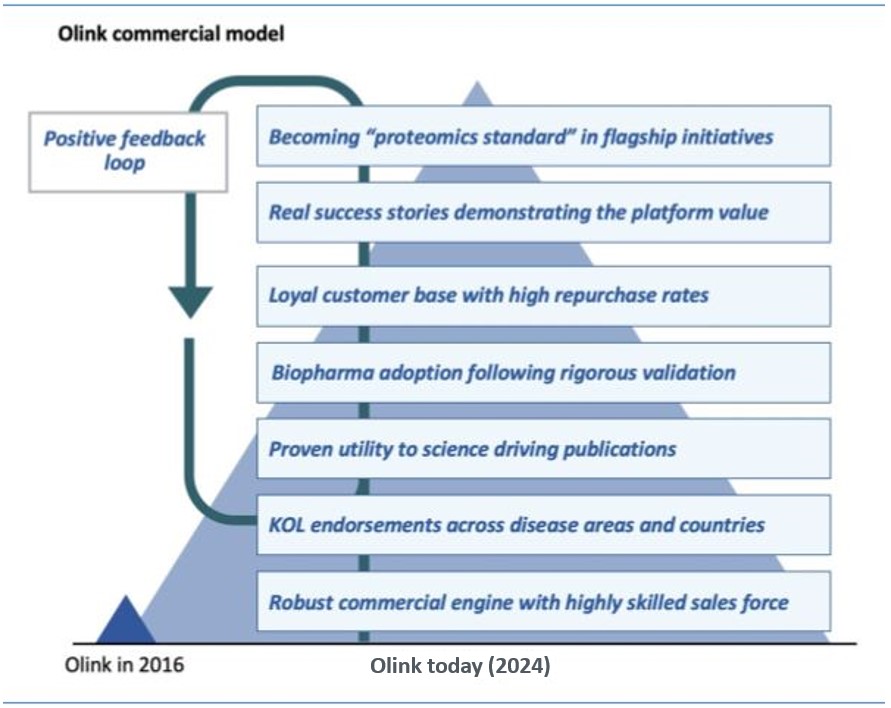
Figure 10. Illustration of Olink’s commercial model and maturation since inception.
/65
Our commercial strategy is focused on driving the adoption of our platform in the research community and expanding our customer base. At the same time, we believe our existing customer relationships are becoming more strategic in nature and that we therefore will be able drive an increasing adoption of our platform with our existing customers. This will require an emphasis on external installations within academic and biopharmaceutical companies’ core facilities, as well as CROs, as well as expanding our portfolio of relevant products and services. In addition to our three product lines, including NGS-readout-based Explore, Target, and Focus, and our purpose-built qPCR readout platform optimized for our Target, Focus and Flex, Olink Signature Q100. We also began taking orders for Olink Flex. We believe Olink Signature Q100 is making our Target and Focus products much more accessible to approximately 4,500 addressable proteomics labs, which combined with the more than 8,900 addressable Illumina systems that we will be able to access with Explore, will make it easier for customers to adopt our platform, allowing us to scale at a faster rate.
Olink launched two additional products in 2022. In October we announced the launch of Olink Insight, an open-access online portal that supports users in understanding and interpreting their proteomics data. We designed Olink Insight to work with Olink data, offering a range of data visualization options that are precise, easy to interpret, and provide an excellent overview of complex data sets, all to accelerate the time it takes for scientists' to generate actionable insights from their data.
In November 2022 we launched Olink Flex, which targets low-plex use-cases running on Olink Signature and Biomark HD, and is a fully flexible made-to-order product for selecting and combining proteins using absolute quantification in one biomarker panel. Using an online panel builder in Olink Insight, customers can freely pick and choose 15 to 21 pre-validated assays from roughly 200 human protein biomarkers covering major biological pathways. We believe that Olink Flex represents a significant innovation in the low-plex market that will make the PEA technology even more accessible for scientists and can serve as an entry level product for modern multi-plex proteomics.
In mid 2023 we expanded our Explore Kit offering through the launch of Olink Explore HT, which targets high-plex use-cases running on next generation sequencing (NGS) with a substantially simplified workflow offering four times higher throughput than its predecessor. Explore HT comprises of 5400 assays, up from 3000 assays with the former generation of Explore, Explore 3072.
Although our strategic focus will be on external installations, we plan to continue to offer our services and invest in our Analysis Service labs. We operate Analysis Service labs in Uppsala, Sweden and Waltham, Massachusetts, from which we support our customers from sample into data out with services including study design and consultation, sample prep, assay execution, data processing, and quality control. In addition, we offer Analysis Service through a third-party service provider in China.
Our commercial and business development teams are consistently developing structures and commercial models designed to lower the barriers of adoption for our customers. In most countries, working with academic or governmental institutions requires us to participate in a tender process or obtain grant applications. These processes require us to support the customer with the necessary documentation, both for our kit products and Analysis Service offerings.
Our global direct sales and marketing efforts are targeted at the PIs, research scientists, department heads, research laboratory directors and core facility directors at leading academic institutions, biopharmaceutical companies and publicly and privately-funded research institutions that control the buying decision. Most importantly, we work closely with many of the most influential KOLs across multiple disease areas and they are our strongest supporters and promoters. These close relationships facilitate the testing of new concepts, generation of more proof points, and the increase in groundbreaking scientific research in proteomics based on PEA, which is then often used as the basis for our marketing activities.
In addition to fostering close relationships within the proteomics scientific community, we increase awareness of our products among our target customers through direct sales calls, trade shows, seminars and webinars, academic conferences, web presence, social media and other forms of internet marketing. We also provide education and training resources, both online and in person.
Manufacturing and Supply Chain
Our manufacturing and supply chain operations are responsible for sourcing the antibodies and other reagents we use in our kit products, as well as the instrumentation required to operate our high-throughput Analysis Service labs.
Part of the antibodies we use in our kit products are sourced from carefully evaluated and approved third- party suppliers. With the acquisition of Agrisera AB, we have taken steps to transition our library towards more in-house developed antibodies. We produce and source our antibodies internally through our facility based out of Umeå, Sweden. These manufacturing operations include: in-house breeding of rabbits, immunization of antigens, and generation of antibodies by affinity purification. As our technology relies on matched pairs of antibodies, we require high-quality antibodies to develop and manufacture our products. The more antibodies required to bind to a protein for identification and read-out, the more
/66
difficult it will be to develop such assays. However, we do not anticipate that many, if any, proteins will require a third antibody for identification and detection and therefore do not consider this a constraint for growing our library or our product development and supply chain going forward.
We obtain some of the components of our kit products from third-party suppliers. While some of these components are sourced from a single supplier, we have qualified second sources for most, but not all, of our critical components and reagents. The loss of any of these suppliers could potentially harm Olink. We seek to mitigate disruption in the supply of a critical component by seeking alternative suppliers and maintaining buffer inventory.
For further discussion of the risks relating to our third-party suppliers, see the section titled “Risk Factors - Risks Related to Our Dependence on Third Parties.”
The reagents used for our kit products or our own Analysis Service labs are manufactured and assembled in Uppsala, Sweden. These manufacturing operations include: reagent formulation, assay formulation, vial- and primer plate filling, kit assembly and packaging as well as analytical and functional quality control testing. We further utilize our lab in Umeå, Sweden for upstream related production and R&D activities related to the manufacturing of our reagent kits.
The instrumentation required to operate our Analysis Service labs is sourced directly from the equipment where we have long-standing relationships.
In June of 2021 we launched our Olink Signature platform, a purpose-built qPCR-based readout platform optimized for running our current and future Target and Focus products, with shipments beginning in the fall of 2021. The instrument is manufactured in Singapore by our OEM-partner.
Seasonal buying patterns of our customers
Customers make significant purchases of our products and services during the fourth quarter of the calendar year, leading to significant concentration of our revenue during this period; historically in excess of 40% of total yearly revenues. This concentration of selling and fulfillment activity can lead to volatility in our reported financial results in the event that revenue recognition of a significant amount of customer orders is pushed into the following year.
Competition
The life science tools space is highly dynamic, with emerging technologies consistently challenging the market position of the more established solutions. In particular, the proteomics market can be characterized as competitive, comprising both well-established legacy technologies and emerging earlier-stage technologies, and with nascent market segments where we do not have an established competition yet.
Intellectual property, market adoption, market perception, customer and KOL relationships, and product quality and performance are essential qualities that differentiate competitors in this market. We classify our current and potential competitors in our three market segments, high-plex, mid-plex and low-plex, where we think their value propositions are most relevant. Established companies with relevant protein detection and quantification technologies include Quanterix Corporation (low-plex), Meso Scale Diagnostics LLC (low- and mid-plex), Luminex Corporation (low- and mid-plex), and SomaLogic, Inc. (high-plex) (Standard Bio Tools has acquired), as well as established proteomics technologies, such as ELISA (low-plex) and mass spectrometry (primarily high-plex), offered by multiple well-known tools providers. In addition, products offered or potentially offered by a number of earlier-stage companies, such as Alamar, Seer, Inc.,, Spear Bio Nautilus Biotechnology, Inc., Quantum-Si Incorporated and Encodia, are also part of the competitive landscape and we believe their emerging technologies are primarily targeting the high-plex segment.
Our commercial opportunity could be reduced if our competitors develop and commercialize products or services that offer better performance or are more convenient and cost-effective to use than our products or services. As a result, a key priority is to continue to invest in driving the technological evolution of PEA as well as to continue to invest in lowering barriers of adoption in the proteomics market in order to accelerate our market position. Equally important, we plan to continue investing in the proteomics scientific community to further develop successful customer stories that demonstrate the value PEA brings to the field of proteomics. We believe we are substantially differentiated from our competitors when considering multiple competitive factors that in combination substantially benefit our customers, including:
•Performance properties, such as specificity, sensitivity, and precision;
•Actionability and clinical utility of the research the technologies enable;
•Scalability by having the ability to support customers from discovery to clinical decision making;
•Accessibility and ease-of-use of underlying detection platforms in the market;
/67
•Data quality and analysis;
•Cost of necessary instrumentation and consumables; and
•Customer service and support.
Intellectual Property
Our success depends in part on our ability to obtain and maintain intellectual property protection for our products and technology. We utilize a variety of intellectual property protection strategies, including patents, trademarks, trade secrets and other methods of protecting proprietary information.
As of December 31, 2023, worldwide we owned or in-licensed 44 issued or allowed patents across ten patent families and 47 pending patent applications across nine patent families. The patent portfolio broadly covers four themes; essential concepts of the overall PEA technology, granted in the US and worldwide, and expiring from 2032 to 2034; how our kit products are designed and manufactured, pending and granted in the US and worldwide, and expiring from 2031 to 2036 or 2042 if pending applications are ultimately granted; sample preparation and workflow, pending as applications in a number of jurisdictions and estimating expiry in 2041, and data analysis, pending in the US and Europe with expected expiry in 2042 and 2043 if granted. A number of our patents and patents applications are for inventions relating to products and methods not currently implemented in our own products.
We also license additional patents on a non-exclusive and/or territory restricted basis. Patent rights generally have a term of twenty years from the date in which they were filed. We own registered trademarks on OLINK, and our logotype in the United States and worldwide, and have pending trademark applications for further names of products and services.
We intend to pursue additional intellectual property protection to the extent we believe it would be beneficial and cost- effective. We cannot provide any assurance that any of our current or future patent applications will result in the issuance of patents, or that any of our current or future issued patents will effectively protect any of our products or technology from infringement or prevent others from commercializing infringing products or technology.
For further discussion of the risks relating to intellectual property, see the section titled “Risk Factors - Risks Related to Intellectual Property”.
Government Regulation
Our focus is on the discovery of biomarkers that our partners use to improve the speed and success of their drug discovery efforts; however, we ourselves are not currently involved in drug discovery, nor do we manufacture any pharmaceutical products, or conduct any clinical trials. As such, while we are subject to a number of regulations, such as those governing our laboratory facilities as well as regulations that apply to businesses in the private sector generally, we are not subject to many of the types of regulations that ordinarily apply to companies in the life sciences, biotechnology and pharmaceutical sectors and industries. However, we believe that the long-term success of our business depends, in part, on our partners’ ability to successfully develop and sell products using the biomarkers that we discover. The regulations that govern our pharmaceutical and biotechnology partners are those we therefore believe have the most significant impact on our business.
Government authorities in the United States, at the federal, state and local level, and in the European Union and other countries and jurisdictions, extensively regulate, among other things, the research, development, testing, manufacturing, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of pharmaceutical products, including biological products, such as those that our partners develop. The processes for obtaining marketing approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
Our partners are and will be subject to a variety of regulations in applicable jurisdictions governing, among other things, clinical studies and any commercial sales and distribution of their products. Regardless of whether our partners obtain Food and Drug Administration (FDA) or European Union (EU) approval for a product, they must obtain the requisite approvals from regulatory authorities in other countries prior to the commencement of clinical studies or marketing of the product in those countries. The requirements and process governing the conduct of clinical studies, product licensing, coverage, pricing and reimbursement vary from country to country.
/68
FDA
In the United States, medical devices are subject to extensive regulation by the FDA, under the Federal Food, Drug, and Cosmetic Act (FDC Act), and its implementing regulations, and other federal and state statutes and regulations. The laws and regulations govern, among other things, medical device development, testing, labeling, storage, premarket clearance or approval, advertising and promotion and product sales and distribution. To be commercially distributed in the United States, medical devices must receive from the FDA prior to marketing, unless subject to an exemption, either approval of a premarket approval (PMA) (for most Class III devices), clearance of a 510(k) premarket notification or classification pursuant to a de novo submission.
IVDs are types of medical devices that can be used in the diagnosis or detection of diseases, conditions or infections, including, without limitation, the presence of certain chemicals, genetic information or other biomarkers. Predictive, prognostic and screening tests, such as carrier screening tests, can also be IVDs. A subset of IVDs is known as analyte- specific reagents (ASRs). ASRs consist of single reagents, and are intended for use in a diagnostic application for the identification and quantification of an individual chemical substance in biological specimens. ASRs are medical devices, but most are exempt from 510(k) review. As medical devices, ASRs have to comply with some Quality System Regulation (QSR) provisions and other device requirements, such as establishment registration, device listing and medical device reporting.
The FDC Act classifies medical devices into one of three categories based on the risks associated with the device and the level of control necessary to provide reasonable assurance of safety and effectiveness. Class I devices are deemed to be low risk and are subject to the fewest regulatory controls. Many Class I devices are exempt from FDA premarket review requirements. Class II devices, including some software products to the extent that they qualify as devices, are deemed to be moderate risk, and generally require clearance through the premarket notification, or 510(k) clearance, process in order to be commercially distributed.
Class III devices are generally the highest risk devices and are subject to the highest level of regulatory control to provide reasonable assurance of the devices’ safety and effectiveness. Class III devices typically require approval of a PMA by the FDA before they are marketed. A clinical study is almost always required to support a PMA application and is sometimes required for 510(k) clearance. All clinical studies of investigational devices must be conducted in compliance with any applicable FDA and Institutional Review Board requirements. Devices that are exempt from FDA premarket review requirements must nonetheless comply with general post-market controls as described below, unless the FDA has chosen to exercise enforcement discretion and not regulate them.
510(k) clearance pathway: To obtain 510(k) clearance, a manufacturer must submit a premarket notification demonstrating to the FDA’s satisfaction that the proposed device is substantially equivalent to a previously 510(k)-cleared device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for submission of PMA applications. The previously cleared device is known as a predicate. The FDA’s 510(k) clearance pathway usually takes from three to 12 months, but it can take longer, particularly for a novel type of product.
PMA pathway: The PMA pathway requires proof of the safety and effectiveness of the device to the FDA’s satisfaction. The PMA pathway is costly, lengthy and uncertain. A PMA application must provide extensive preclinical and clinical trial data as well as information about the device and its components regarding, among other things, device design, manufacturing and labeling. As part of its PMA review process, the FDA will typically inspect the manufacturer’s facilities for compliance with QSR requirements, which impose elaborate testing, control, documentation and other quality assurance procedures. The PMA review process typically takes one to three years but can take longer.
De novo pathway: If no predicate device can be identified, a device is automatically classified as a Class III device, requiring a PMA application. However, the FDA can reclassify, or use “de novo classification,” for a device for which there was no predicate device if the device is low or moderate risk. The FDA will identify “special controls” that the manufacturer must implement, which often include labeling and other restrictions. Subsequent applicants can rely on the de novo product as a predicate for a 510(k) clearance. The de novo route is less burdensome than the PMA process. A device company can ask the FDA at the outset if the de novo route is available and submit the application as one requesting de novo classification. The de novo route has been used for many IVD products.
/69
Post-market general controls. After a device, including a device exempt from FDA premarket review, is placed on the market, numerous regulatory requirements apply. These include the QSR, labeling regulations, registration and listing, the Medical Device Reporting regulation (which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur) and the Reports of Corrections and Removals regulation (which requires manufacturers to report recalls and field actions to the FDA if initiated to reduce a risk to health posed by the device or to remedy a violation of the FDC Act).
The FDA enforces these requirements by inspection and market surveillance. If the FDA finds a violation, it can institute a wide variety of enforcement actions, ranging from an untitled or public warning letter to more severe sanctions such as fines, injunctions and civil penalties; recall or seizure of products; operating restrictions and partial suspension or total shutdown of production; refusing requests for 510(k) clearance or PMA approval of new products; withdrawing 510(k) clearance or PMAs already granted; and criminal prosecution.
Research Use Only
An RUO product is one that is not intended for clinical diagnostic use and must be labeled “For Research Use Only. Not for use in diagnostic procedures.” Products that are intended for research use only and are properly labeled as RUO are exempt from compliance with the FDA requirements discussed above, including the approval or clearance and most QSR requirements. A product labeled RUO but intended to be used diagnostically may be viewed by the FDA as adulterated and misbranded under the FDC Act and is subject to FDA enforcement activities. The FDA may consider the totality of the circumstances surrounding distribution and use of an RUO product, including how the product is marketed, when determining its intended use. In November 2013, the FDA issued a guidance document entitled “Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only” (RUO Guidance) which highlights the FDA’s interpretation that distribution of RUO products with any labeling, advertising or promotion that suggests that clinical laboratories can validate the test through their own procedures and subsequently offer it for clinical diagnostic use as a laboratory developed test is in conflict with RUO status. The RUO Guidance further articulates the FDA’s position that any assistance offered in performing clinical validation or verification, or similar specialized technical support, to clinical laboratories, conflicts with RUO status.
Laboratory-developed tests (LDTs)
LDTs have generally been considered to be tests that are designed, developed, validated and used within a single laboratory. The FDA takes the position that it has the authority to regulate such tests as medical devices under the FDC Act. The FDA has historically exercised enforcement discretion and has not required clearance or approval of LDTs prior to marketing. In addition, the New York Clinical Laboratory Evaluation Program separately approves certain LDTs offered to New York State patients.
On October 3, 2014, the FDA issued two draft guidance documents regarding oversight of LDTs. These draft guidance documents proposed more active review of LDTs. The draft guidance documents have been the subject of considerable controversy, and in November 2016, the FDA announced that it would not be finalizing the 2014 draft guidance documents. On January 13, 2017, the FDA issued a discussion paper which laid out elements of a possible revised future LDT regulatory framework, but did not establish any regulatory requirements.
On September 29, 2023, the FDA announced a proposed rule that would explicitly state that IVDs are devices under the FDC Act and would phase out FDA's general enforcement discretion with respect to LDTs and regulate LDTs as IVDs such that LDTs would be considered devices. The proposed rule was published on October 3, 2023 and the notice and comment period closes on December 4, 2023. At the end of the notice and comment period, the FDA may decide to abandon the rule, issue a final rule as proposed or issue a final rule with modifications.
The FDA’s efforts to regulate LDTs have also prompted the drafting of legislation governing diagnostic products and services that sought to substantially revamp the regulation of both LDTs and in vitro diagnostics, or IVDs. Congress may act to provide further direction to the FDA on the regulation of LDTs.
Further, certain additional healthcare regulations may apply if we expand into new product lines or services, such as federal and state fraud and abuse, transparency and health information privacy and security laws and state clinical laboratory requirements, among others.
/70
Privacy Laws
We also are or may become subject to data protection and privacy laws and regulations in the jurisdictions in which we are established, have partners, or sell or market our services. Processing of personal data, including health related information, is increasingly subject to legislation and regulations in numerous jurisdictions around the world, including the EU’s General Data Protection Regulation (GDPR), Canada’s Personal Information Protection and Electronic Documents Act (PIPEDA) and the analogous provincial laws, the Health Insurance Portability and Accountability Act of 1996 (HIPAA) in the United States, California's Confidentiality of Medical Information Act (CMIA), and the California Consumer Privacy Act (CCPA) among many others. Our regulatory obligations in foreign jurisdictions could harm the use or cost of our solution in international locations as data protection and privacy laws and regulations around the world continue to evolve.
In Europe we are subject to the GDPR (Regulation (EU) 2016/679) and related applicable data protection and privacy laws of the member states of the European Economic Area, in relation to our processing and other use of personal data (i.e. data relating to an identifiable living individual) as part of our provision of services to customers and in connection with the administration and operation of our business. The GDPR is wide-ranging in scope and imposes numerous additional requirements on companies that process personal data, including imposing special requirements in respect of the processing of health and other sensitive data. The GDPR imposes accountability obligations requiring data controllers and processors to maintain a record of their data processing and implement policies and procedures as part of its mandated privacy governance framework. It also requires data controllers to be transparent and disclose to data subjects how their personal data will be used; establishes rights for individuals with respect to their personal data, including rights of access and deletion in certain circumstances; imposes limitations on retention of personal data; establishes mandatory data breach notification requirements; and sets higher standards for data controllers to demonstrate that they have obtained valid consent for certain data processing activities. For more information on GDPR risks, please refer to the section entitled, "ITEM 3 - KEY INFORMATION - D. Risk Factors" in this Form 20-F.
EU Member States may introduce further conditions, including limitations which could limit our ability to collect, use and share personal data (including health and medical information), or could cause our compliance costs to increase. In addition, the GDPR imposes strict rules on the transfer of personal data out of the EU/UK to third countries deemed to lack adequate privacy protections (including the U.S.), unless an appropriate safeguard specified by the GDPR is implemented, such as the Standard Contractual Clauses (SCCs) approved by the European Commission, or a derogation applies. The Court of Justice of the European Union (CJEU) recently confirmed in its judgment in the "Schrems II" case (Case C‑311/18) in July 2020 that the SCCs remain a valid mechanism for transfers of personal data to third countries. However, the CJEU also ruled that transfers made pursuant to the SCCs and other alternative transfer mechanisms need to be analyzed on a case-by- case basis to ensure EU standards of data protection are met in the jurisdiction where the data importer is based, and there continue to be concerns about whether the SCCs and other mechanisms will face additional challenges. European regulators have issued guidance following the CJEU ruling that imposes significant diligence requirements on transferring data outside the EEA, including under an approved transfer mechanism. This guidance requires an “essential equivalency” assessment of the laws of the destination country. If essentially equivalent protections are not available in the destination country, the exporting entity must then assess if supplemental measures can be put in place that, in combination with the chosen transfer mechanism, would address the deficiency in the laws and ensure that essentially equivalent protection can be given to the data. Complying with this guidance will be expensive and time consuming and, in the worst case scenario, may ultimately prevent us from transferring personal data to jurisdictions outside the EEA where essentially equivalent protection of personal data is not available, which would cause significant business disruption. Like many other businesses, until the legal uncertainties regarding how to legally continue transfers pursuant to the SCCs and other mechanisms are settled, we will continue to face uncertainty as to whether our efforts to comply with our obligations under the GDPR will be sufficient. This and other future developments regarding the flow of data across borders could increase the complexity of transferring personal data across borders in some markets and may lead to governmental enforcement actions, litigation, fines and penalties or adverse publicity, which could have an adverse effect on our reputation and business. That said, as far as transfers of personal data from the EU to the US are concerned, on 10 July 2023, the EU and US announced that they had reached an agreement in principle on a new deal to allow personal data to transfer from the EEA to the US to try and resolve the uncertainty created by the above decision (known as the DPF) and to replace the previous EU-US Privacy Shield framework which was invalidated in the "Schrems II" case. The US is now subject to an adequacy decision from the European Commission which is now in place.
The GDPR creates sanctions for breach of data protection obligations with potential fines that are significant: up to the greater of €20 million or 4% of total global annual turnover. The authorities have shown a willingness to impose significant fines and issue orders preventing the processing of personal data on non- compliant businesses. Moreover, individuals can claim damages resulting from infringement of the GDPR and other European data protection laws. The GDPR also introduces the right for non-profit organizations to bring claims on behalf of data subjects. In addition to the foregoing, a
/71
breach of the GDPR or other applicable privacy and data protection laws and regulations could result in regulatory investigations, reputational damage, orders change our use of data, enforcement notices, or potential civil claims including class action type litigation.
In addition, as of January 1, 2021, the GDPR was replicated in UK law as the ‘UK GDPR’, but there may be further developments about the regulation of particular issues with which we will be required to comply if there is subsequently divergence between UK and EU regulation of data protection.
The risk of our Company being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by applicable regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. The shifting compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and/or reporting requirements increases the possibility that a healthcare company may run afoul of one or more of the requirements.
Compliance with data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure by us or our collaborators and third-party providers to comply with data protection laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), private litigation and/or adverse publicity and could negatively affect our operating results and business. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend, could result in adverse publicity and could have a material adverse effect on our business, financial condition, results of operations and prospects.
Additional Regulation
In addition to the foregoing, supranational, national, state and federal U.S. and European laws regarding environmental protection and hazardous substances affect our business. These and other laws govern our use, handling and disposal of various biological, chemical and radioactive substances used in, and wastes generated by, our operations. If our operations result in contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and governmental fines. We believe that we are in material compliance with applicable environmental laws and that continued compliance therewith will not have a material adverse effect on our business. We cannot predict, however, how changes in these laws may affect our future operations.
Anti-Corruption Laws
We are subject to the U.S. Foreign Corrupt Practices Act of 1977, as amended (FCPA), the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, and other state and national anti-bribery and anti- money laundering laws in countries in which we conduct activities, such as the UK Bribery Act 2010 and the UK Proceeds of Crime Act 2002, collectively, Anti-Corruption Laws.
Among other matters, such Anti-Corruption Laws prohibit corporations and individuals from directly or indirectly paying, offering to pay or authorizing the payment of money or anything of value to any foreign government official, government staff member, political party or political candidate, or certain other persons, in order to obtain, retain or direct business, regulatory approvals or some other advantage in an improper manner. Such Anti-Corruption Laws may also include commercial bribery and other prohibitions that make it illegal for our employees and contractors to give or receive money or anything of value in an improper manner, regardless of whether a foreign official is involved. We may also be held liable for the acts of our third party agents under the FCPA, the UK Bribery Act 2010 and other Anti-Corruption Laws. In the healthcare sector, anti-corruption risks can also arise in the context of improper interactions with doctors, KOLs and other healthcare professionals who work for state-affiliated hospitals, research institutions or other organizations or in relation to healthcare providers.
/72
C.Organizational Structure
Below is a list of the significant subsidiaries of Olink, including our ownership percentage, its year of formation and its jurisdiction. These subsidiaries were established to allow us to conduct commercial and clinical operations and expand our operations globally.
| Share of common shares owned by the Company (%) | |||||||||||||||||
| Name | Principle Activities | Year of formation | Country of registration and operation | 2023 | 2022 | ||||||||||||
| Olink Finance AB | Cash management | 2018 | Sweden | 100% | 100% | ||||||||||||
| Olink OldCo AB | Other operational activities | 2016 | Sweden | 100% | 100% | ||||||||||||
| Olink Proteomics AB | Sales, production, and research & development | 2015 | Sweden | 100% | 100% | ||||||||||||
| Agrisera AB | Production, and research & development | 1985 | Sweden | 100% | 100% | ||||||||||||
| Olink Proteomics Inc. | Sales of services and distribution services | 2015 | USA | 100% | 100% | ||||||||||||
| Olink Proteomics Ltd | Marketing coordination and sales services | 2015 | UK | 100% | 100% | ||||||||||||
| Olink Proteomics B.V | Marketing coordination and sales services | 2016 | Netherlands | 100% | 100% | ||||||||||||
| Olink Proteomics GmbH. | Marketing coordination and sales services | 2018 | Germany | 100% | 100% | ||||||||||||
| Olink Proteomics KK | Marketing coordination and sales services | 2019 | Japan | 100% | 100% | ||||||||||||
| Olink Biotech (Shanghai) Co., Ltd | Distribution, marketing coordination and sales services | 2020 | China | 100% | 100% | ||||||||||||
| Olink Proteomics SAS | Marketing coordination and sales services | 2022 | France | 100% | 100% | ||||||||||||
| Olink Proteomics SG Pte. Ltd. | Marketing coordination and sales services | 2023 | Singapore | 100% | N/A | ||||||||||||
D.Property, Plants and Equipment
Our corporate headquarters, research and development facilities and manufacturing distribution centers and our largest Analysis Service lab are located in Uppsala, Sweden, where we lease approximately 57,000 square feet of space under leases has expired in December 31, 2023. We also lease approximately 73,000 square feet of office and laboratory space for our new headquarters in Uppsala, Sweden and which we have taken occupancy in July 2023. This lease expires on March 31, 2033. We lease approximately 32,000 square feet of office and laboratory space in Waltham, Massachusetts. This lease expires on May 31, 2029. In Shanghai, China we lease approximately 2,300 square feet, pursuant to a lease expiring on April 15, 2024. In Umeå, Sweden, Olink leases approximately 13.000 square feet of laboratory and office expiring March 31, 2027, and approximately 15,000 square feet of supplemental facility space, expiring April 30, 2030. In Umeå, Sweden, Olink also owns an office/laboratory and related buildings (on leased ground) of approximately 4,575 square feet in size. In our remaining geographies, we lease approximately 2,950 of combined square feet, with various expiration dates.
/73
Except as noted above, we do not own any real property and believe that our current facilities are sufficient to meet our ongoing needs and that, if we require additional space, we will be able to obtain additional facilities on commercially reasonable terms.
ITEM 4A. UNRESOLVED STAFF COMMENTS
None.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
The following “Operating and Financial Review and Prospects” should be read together with the information in our financial statements and related notes included elsewhere in this Annual Report. The following discussion is based on our financial information prepared in accordance with the IFRS Accounting Standards, as issued by the International Accounting Standards Board, or IASB, which may differ in material respects from generally accepted accounting principles in other jurisdictions, including U.S. GAAP. The following discussion includes forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those described in “Risk Factors” and elsewhere in this Annual Report. Please also see “Special Note Regarding Forward-Looking Statements.”
Overview
Our purpose is to enable and accelerate the field of proteomics by providing a platform of products and services, developed with key opinion leaders (KOLs), that are deployed across major biopharmaceutical companies and leading clinical and academic institutions, to deepen the understanding of real-time human biology and drive 21st century healthcare through actionable and impactful science.
Our dedication to customer satisfaction and quality has enabled us to expand our existing customer base from inception in 2016. Revenues from our original customer accounts that we obtained in 2016 have grown at an average annual growth rate of 31%. These original customer accounts we've had since 2016 represented approximately 23% of our revenues for the year ended December 31, 2023.
Our customers primarily include academic, government, biopharmaceutical, biotechnology and other institutions focused on life science research. Our revenue is principally generated from two segments, Kit and Service. Kit revenues refer to the sale of our panels directly to customers that run the kit and analysis in their own labs. During the year ended December 31, 2023 and year ended December 31, 2022, sales to academic institutions and core labs represented approximately 55% and 45% of our revenues, respectively. Sales to biopharmaceutical companies represented the remaining 45% and 56% of our revenues, respectively. We operate a global direct sales model across all our regions (Americas, EMEA and APAC) and customer segments. As of December 31, 2023, our commercial team was comprised of 208 employees, with an emphasis on the Americas region. Sales within the Americas accounted for approximately 46% of revenues during the year ended December 31, 2023 and approximately 48% of our revenues during the year ended December 31, 2022.
/74
A.Operating Results Financial Operations Overview
The following table summarizes our results of operations for the periods presented:
| Amounts in thousands of USD | Year ended December 31, 2023 | Year ended December 31, 2022 | ||||||
| Revenue | $ | 169,597 | $ | 139,848 | ||||
| Cost of revenue | (55,136) | (45,349) | ||||||
| Gross profit | 114,461 | 94,499 | ||||||
| Selling expenses | (54,479) | (44,673) | ||||||
| Administrative expenses | (75,648) | (54,274) | ||||||
| Research and development expenses | (34,183) | (26,345) | ||||||
| Other operating income | 2,243 | 4,464 | ||||||
| Other operating expense | $ | (2,670) | $ | (4,273) | ||||
| Operating loss | (50,276) | (30,602) | ||||||
| Interest income | 6,514 | 1,159 | ||||||
| Interest expense | (858) | (531) | ||||||
| Foreign exchange gain | 4,137 | 14,059 | ||||||
| Other financial income/(expense) | 578 | 508 | ||||||
| Loss before tax | (39,905) | (15,407) | ||||||
| Income tax benefit | 8,305 | 2,556 | ||||||
| Net loss for the period (Attributable to shareholders of the Company) | $ | (31,600) | $ | (12,851) | ||||
| Other comprehensive income/(loss): | ||||||||
| Items that may be reclassified to profit or loss: | ||||||||
| Exchange differences from translation of foreign operations | 16,293 | (60,289) | ||||||
| Other comprehensive income/(loss) for the, period, net of tax | 16,293 | (60,289) | ||||||
| Total comprehensive loss for the period, net of tax | $ | (15,307) | $ | (73,140) | ||||
| Total comprehensive loss for the period (Attributable to owners of the Company) | $ | (15,307) | $ | (73,140) | ||||
| Basic and diluted loss per share | $ | (0.25) | $ | (0.11) | ||||
Year Ended December 31, 2023 Compared to Year Ended December 31, 2022 Revenue
We principally derived our revenues from the sale of our biomarker panels, either as a kit-product or by providing analysis and ancillary services for customers that prefer outsourced proteomics analysis. Overall, 2023 revenue for the year ended December 31, 2023 was $169.6 million compared to $139.8 million for the year ended December 31, 2022 This increase of $29.7 million, or 21.3%, was mainly due to expansion of Kit business and the continued roll out of our Explore offering (Explore HT), coupled with accelerated growth for our Target portfolio on the back of the Signature launch in late 2021.
Cost of revenue
/75
Cost of revenue primarily consists of manufacturing costs incurred in the production process including personnel and related costs; costs of component materials; depreciation of plant & machinery. equipment & tools and leases; manufacturing overhead; delivery costs and allocated costs including facilities and information technology. In addition, cost of revenue includes royalty costs for licensed technologies included in our products, provisions for slow-moving and obsolete inventory.
Cost of revenue for the year ended December 31, 2023 was $55.1 million compared to $45.3 million for the year ended December 31, 2022. The increase of $9.8 million, or 21.6%, was mainly volume driven due to growing Kit business, but also due to an increase in consumable expenses as well as higher royalty fees.
Gross Profit/Gross Profit Percentage
Gross profit is calculated as revenue less cost of revenue. Gross profit percentage is gross profit expressed as a percentage of revenue. We expect our future gross profit and gross profit percentages to fluctuate from period to period. Future gross profit and gross profit percentages will depend on a variety of factors, including: market conditions that may impact our pricing; sales mix changes among kit, instruments and services; product mix changes between established products and new products; excess and obsolete inventories; royalties; and our cost structure for manufacturing operations relative to volume.
As we seek to increase our production and distribution platform, we may incur incremental costs that potentially will reduce the gross profit percentage in certain periods.
Gross profit for the year ended December 31, 2023 was $114.5 million compared to $94.5 million for the year ended December 31, 2022. The increase of $20.0 million, or 21.1%, was due to year over year revenue growth. The gross profit percentage decreased from 67.6% to 67.5%, represents a decrease in gross profit percentage of (0.1)%, which is primarily explained by an increase in logistics expenses, such as shipping and freight. Kit revenue as percentage of total revenues increased from 39.4% for the year ended December 31, 2022 to 51.6% for the year ended December 31, 2023.
Operating Expenses
Selling Expenses
Selling expense primarily consists of costs related to the selling and marketing of our products, including sales incentives and advertising expenses and costs associated with our global commercial team. Selling expenses include costs associated with the commercial team; recruiting services; administrative services; public relations and communication activities; marketing programs and trade show appearances; travel; customer service costs; and allocated costs, including facilities and information technology; and fees for third-party providers of administrative services, including press relations and communication services; security, reception, and recruiting.
Selling expenses for the year ended December 31, 2023 was $54.5 million, or 32.1% of our total revenue, compared to
$44.7 million, or 31.9% of our total revenue, for the year ended December 31, 2022. This increase of $9.8 million, or 22.0%, was primarily driven by higher employee benefits expense, consisting of wages, salaries, social security and pension costs to employees in selling functions. This has been driven by our ongoing effort to build out our global commercial capabilities.
Administrative Expenses
Administrative expenses include costs associated with our finance, accounting, legal, human resources, communications, and administrative personnel; facility-related costs; and intellectual property fees for the registration and maintenance of our patents. We anticipate that our administrative expenses will increase in the future as we grow our support functions in line with our planned growth. We also anticipate increased expenses associated with being a public company in the United States, including costs related to audit, legal, regulatory and tax-related services associated with maintaining compliance with U.S. exchange listing and SEC requirements, director and officer insurance premiums, and investor relations costs. In particular, we will incur additional accounting expenses to comply with the Sarbanes-Oxley Act in the United States that require us to test the effectiveness of our internal controls over financial reporting.
Administrative expenses for the year ended December 31, 2023 totaled $75.6 million, or 44.6% of our total revenue, compared to $54.3 million, or 38.8% of our total revenue, for the year ended December 31, 2022. The increase of $21.4 million, or 39.4% is reflective of the overall growth of the business, which includes deal related and capital markets related expenses.
/76
Research and Development Expenses
Research and Development expenses associated with our research and development functions, primarily located in Uppsala and Umeå, Sweden. Expenses include costs of employee benefit expenses of our R&D personnel, R&D facility-related costs, recruitment, administrative services and allocated costs including facilities and information technology, and intellectual property fees for the registration and maintenance of our patents.
We deploy a substantial portion of our resources on developing new products and solutions. Our research and development efforts are focused on identifying and developing new biomarker expressions through our Affinity program, improving the performance in existing products and developing new product lines and features.
We plan to continue to invest significantly in our research and development efforts, including hiring additional employees, to enhance existing products and develop new products. Our Affinity program is focused on expanding our library of proteins beyond approximately 5,400 that is commercially available to our customers today. The expansion of our library of proteins was further enabled by the acquisition of Agrisera in 2020; that vertically integrated our supply chain and enabled in house antibody production.
Research and Development expenses for the year ended December 31, 2023 totaled $34.2 million, or 20.2% of our total revenue, compared to $26.3 million, or 18.8% of our total revenue, for the year ended December 31, 2022. The increase of $7.8 million, or 29.8%, was driven by $4.2 million higher employee benefits expenses, consisting of wages, salaries, social security, and pension costs to employees in research and development functions. The majority of the external spend within our R&D function was focused on the development of new assays and expansion of our library of protein biomarkers.
Financial Income (Expense)
Interest income relates primarily to interest income received from cash at bank. Our cash at bank has been deposited in cash accounts and therefore generates only a modest amount of interest income. Interest expense relates primarily to interest expense on our outstanding leases.
We also incur foreign exchange gains and losses, mainly related to revaluation of bank balances denominated in foreign currencies, which amounts are recorded as foreign exchange gain/(loss).
Interest income for the year ended December 31, 2023 was $6.5 million, compared to $1.2 million for the year ended
December 31, 2022. The increase is mainly explained by larger interest income on bank deposits due to increased interest rates.
December 31, 2022. The increase is mainly explained by larger interest income on bank deposits due to increased interest rates.
Foreign exchange gain Interest income for the year ended December 31, 2023 was $4.1 million compared to $14.1 million for the year ended December 31, 2022. The decrease is explained by larger interest income on bank deposits and foreign currency gains related to revaluation of bank balances.
Financial expense for the year ended December 31, 2023 was $(0.9) million, compared to $(0.5) million for the year ended December 31, 2022.
Income Taxes
Our tax credit or expense consists of income taxes, with Swedish income taxed at the Swedish tax rate and taxation for other jurisdictions calculated at the rates prevailing in each respective jurisdiction. Income taxes also include the impact of temporary differences which is primarily due to the acquisition accounting for the intangible assets.
Income tax benefit for the year ended December 31, 2023 was $8.3 million compared to a benefit of $2.6 million for the year ended December 31, 2022. The statutory Swedish tax rate was 20.6% during 2022 and 2023.
/77
Segment Information
We report results under two segments: Kit and Service, as further discussed in the Segment Information sections below within Components of Results of Operations and Results of Operations. All other operating segments have been aggregated and are included within the All other segments heading.
Kit Revenues
Kit revenues represented 51.6% of our revenues for the year ended December 31, 2023 compared to 39.4% for the year ended December 31, 2022 and grew 58.8% year over year primarily as a result of the December 2020 launch of our Explore products. We generated an adjusted gross profit percentage of 84.2% on Kit revenues for the year ended December 31, 2023 compared to 88.4% for the year ended December 31, 2022. The growth in FY23 was driven by our continued efforts to expand our footprint of Explore (including Explore HT) customer running our kits in house and continued placements of the Signature instrument that drives Target kit revenues.
Service Revenues
Historically, services have been the main source of our revenue and a key driver of our financial performance. Service revenues represented 38.9% of our revenues for the year ended December 31, 2023 compared to 52.2% for the year ended December 31, 2022 and declined (9.5)% year over year primarily as a result of the shift to Kit business. We generated an adjusted gross profit percentage of 61.4% on Service revenues during the year ended December 31, 2023 compared to 60.1% during the year ended December 31, 2022 which represents an increase in adjusted gross profit percentage of 1.3%.
Year Ended December 31, 2022 Compared to Year Ended December 31, 2021
For a discussion of our consolidated statements of operations for the year ended December 31, 2022 compared to the year ended December 31, 2021, see the section “Item 5. Operating and Financial Review and Prospects” in our Form 20-F for the fiscal year December 31, 2022 filed with the SEC on March 27, 2023.
Non-IFRS Reconciliations
We present the following non-IFRS financial measures because they are used by our management to evaluate our operating performance and formulate business plans. We also believe that the use of these non-IFRS measures facilitates investors’ assessment of our operating performance. We caution readers that amounts presented in accordance with our definitions of Adjusted EBITDA, Adjusted Gross Profit and Adjusted Gross Profit Percentage may not be the same as similar measures used by other companies. Not all companies and Wall Street analysts calculate the non-IFRS measures we use in the same manner. We compensate for these limitations by reconciling each of these non-IFRS measures to the nearest IFRS performance measure, which should be considered when evaluating our performance. Non-IFRS financial measures should not be considered a substitute for our superior to IFRS measures. We encourage you to review our financial information in its entirety and not rely on a single financial measure.
Adjusted EBITDA
We use the non-IFRS measure of Adjusted EBITDA, which we define as profit for the year before accounting for finance income, finance costs, tax, management adjustments, share based compensation expenses, depreciation, and amortization of acquisition intangibles. Management adjustments generally consist of certain cash and non-cash items that we believe are not reflective of the normal course of our business. We identify and determine items to be unique based on their nature and incidence or by their significance. As a result, the composition of these items may vary from year to year.
We present Adjusted EBITDA because we believe this measure can provide useful information to investors and analysts regarding the operational results of the business, as EBITDA is a fairly common metric with which market participants are familiar.
/78
A reconciliation of Adjusted EBITDA to operating loss, the closest line within the IFRS consolidated statement of income and other comprehensive income, is set forth below:
| Amounts in thousands of USD | Year ended December 31, 2023 | Year ended December 31, 2022 | ||||||
| Operating Loss | (50,277) | (30,602) | ||||||
| Add: | ||||||||
| Amortization | 11,029 | 11,212 | ||||||
| Depreciation | 7,971 | 6,114 | ||||||
| EBITDA | (31,277) | (13,276) | ||||||
| Management Adjustments | 14,432 | 1,288 | ||||||
| Share based compensation expenses | 11,060 | 8,047 | ||||||
| Adjusted EBITDA | $ | (5,785) | $ | (3,941) | ||||
Management adjustments for the year ended December 31, 2023 amounted to $14.4 million of costs associated with the January 2023 capital raise and costs associated with the tender offer by Thermo Fisher Scientific Inc. announced on October 17, 2023. The costs associated with the tender offer are attributable specifically to third-party administrative expenses, mainly legal fees accounted to $8.0 million and financial services of $6.3 million. Management adjustments for the year ended December 31, 2022 amounted to $1.3 million in total of costs primarily relating to initial and secondary offering in 2021. Adjusted EBITDA for the year ended December 31, 2023 also includes an add back of $11.1 million of share based compensation expenses associated with our Amended and Restated 2021 Incentive Award Plan.
Adjusted Gross Profit, including Adjusted Gross Profit Percentage
We use the non-IFRS measure of Adjusted Gross Profit, including Adjusted Gross Profit Percentage. We define Adjusted Gross Profit as revenue less cost of revenue, which is then adjusted to remove the impact of depreciation and the impact of material transactions or events that we believe are not indicative of our core operating performance, such as share based compensation expenses and any inventory fair value step up associated with the purchase accounting process that is recorded within cost of revenue, which may or may not be recurring in nature.
We believe that Adjusted Gross Profit, including Adjusted Gross Profit Percentage, provides important information to management and to investors regarding our core profit margin on sales. These are primary profit or loss measures we use to make resource allocation decisions and evaluate segment performance. Adjusted gross profit assists management in comparing the segment performance on a consistent basis for purposes of business decision-making by removing the impact of certain items we believe do not directly reflect our core operations and, therefore, are not included in measuring segment performance.
Reconciliations of Adjusted Gross Profit to gross profit, the most directly comparable IFRS measure, are set forth below:
| Amounts in thousands of USD, unless otherwise stated | Year ended December 31, 2023 | Year ended December 31, 2022 | ||||||
| Revenue | 169,597 | 139,848 | ||||||
| Cost of revenue | (55,136) | (45,349) | ||||||
| Gross Profit | 114,461 | 94,499 | ||||||
| Gross Profit % | 67.5 | % | 67.6 | % | ||||
| Less: | ||||||||
| Depreciation charges | 3,230 | 3,017 | ||||||
| Share based compensation expenses | 646 | 396 | ||||||
| Adjusted Gross Profit | $ | 118,337 | $ | 97,912 | ||||
| Adjusted Gross Profit % | 69.8 | % | 70.0 | % | ||||
/79
Reconciliations of Adjusted Gross Profit to gross profit, the most directly comparable IFRS measure, by segment, are set forth below:
| Amounts in thousands of USD, unless otherwise stated | Year ended December 31, 2023 | Year ended December 31, 2022 | ||||||
| Kit | ||||||||
| Revenue | 87,493 | 55,091 | ||||||
Cost of revenue | (14,946) | (7,131) | ||||||
| Gross profit | $ | 72,547 | $ | 47,960 | ||||
| Gross profit margin | 82.9 | % | 87.1 | % | ||||
| Less: | ||||||||
| Depreciation charges | 887 | 569 | ||||||
| Share-based compensation expenses | 275 | 176 | ||||||
| Adjusted Gross Profit | $ | 73,709 | $ | 48,705 | ||||
| Adjusted Gross Profit % | 84.2 | % | 88.4 | % | ||||
| Service | ||||||||
| Revenue | 66,048 | 73,012 | ||||||
Cost of revenue | (28,191) | (31,776) | ||||||
| Gross profit | $ | 37,857 | $ | 41,236 | ||||
| Gross profit margin | 57.3 | % | 56.5 | % | ||||
| Less: | ||||||||
| Depreciation charges | 2,343 | 2,448 | ||||||
| Share-based compensation expenses | 371 | 220 | ||||||
| Adjusted Gross Profit | $ | 40,571 | $ | 43,904 | ||||
| Adjusted Gross Profit % | 61.4 | % | 60.1 | % | ||||
All other segments | ||||||||
| Revenue | 16,056 | 11,745 | ||||||
Cost of revenue | (11,999) | (6,442) | ||||||
| Gross profit | $ | 4,057 | $ | 5,303 | ||||
| Gross profit margin | 25.3 | % | 45.2 | % | ||||
| Less: | ||||||||
| Depreciation charges | — | — | ||||||
| Share-based compensation expenses | — | — | ||||||
| Adjusted Gross Profit | $ | 4,057 | $ | 5,303 | ||||
| Adjusted Gross Profit % | 25.3 | % | 45.2 | % | ||||
Adjusted gross profit percentage for the year ended December 31, 2023 was 69.8% compared to an adjusted gross profit percentage of 70.0% for year ended December 31, 2022. Adjusted gross profit for the years ended December 31, 2023 and 2022 consists of $3.2 million and $3.0 million, respectively, related to depreciation charges.
Impact of Covid
The COVID-19 pandemic negatively affected parts of our business for longer parts of 2020, in 2021-2022 we observed a lower impact from the COVID-19 pandemic than in 2020, but some regions such as China were more affected by continued shutdowns in 2021 and 2022. Our production and manufacturing facilities are located in Uppsala, Sweden and
/80
Waltham, Massachusetts, and to date we have not experienced any material disruptions in our production or delivery of goods. We increased our inventory level 2020-2021 to be able to work with a higher inventory than we had done historically. Although we saw a reduction in demand due to the lingering effects of the COVID-19 pandemic, we have not observed any significant changes in our underlying customer base. As of December 31, 2023, we concluded that there was no evidence of material changes in the recovery risk of business assets, including deferred tax assets and trade receivables. In light of the total duration of the risk factor and that COVID-19 is still widespread and if a new variant flares up, and the government restrictions may reappear, we continue to follow this closely.
B.Liquidity and Capital Resources
Since our inception, until March 7, 2019, we financed our operations primarily through internally generated cash flows and we did not rely on any material external financing arrangements during this period.
On March 29, 2021, we completed our initial public offering of 13,235,294 ADSs, representing 13,235,294 common shares, at an initial public offering price of $20.00 per share. The net proceeds from the initial public offering were $249.3 million, after deducting the underwriting discounts, net of deferred taxes, and other initial public offering costs associated with the filing. On March 30, 2021, we repaid $65.6 million of outstanding loan facilities plus accrued interest of $1.9 million using the net proceeds from the offering.
On January 18, 2023, the Group initiated a public offering of 5,831,028 American Depositary Shares, each representing one common share of the Group (the “ADSs”), consisting of 4,250,000 ADSs offered by the Company and 1,581,028 ADSs offered by certain selling shareholders of the Group (the “Selling Shareholders”), at a price to the public of $20.00 per ADS. In addition, the Group granted the underwriters a 30-day option to purchase up to an additional 874,654 ADSs from the Company. The Company did not receive any proceeds from the sale of the ADSs by the Selling Shareholders. The offering closed on January 23, 2023 with respect to the initial 4,250,000 ADSs offered by the company and 1,581,028 ADSs/shares offered by the selling stockholders. The option granted to the underwriters closed February 13, 2023 with a total of 760,253 ADSs offered by the company pursuant to the 30-day time period. Total proceeds from the share issue after deducting the underwriting discounts, but before deducting other public offering costs is $95.2 million.
As of December 31, 2023, we had $121.0 million in cash at bank and no outstanding loan balances, compared to $75.1 million and no outstanding loan balances as of December 31, 2022.
Loan Facilities
During the year ended December 31, 2019 the Group entered into a loan facility in the amount of $110 million with Bridgepoint Credit and DNB AB (Publ) as part of the financing of the Olink Acquisition (Facilities). During the year ended December 31, 2020, we amended our debt structure under the existing loan facility with Bridgepoint Credit and DNB AB (Publ), increasing the total commitment under the facilities to $137.6 million. The effective date of the amended agreement was December 23, 2020.
A total of $63.5 million had been drawn down under the term Facility B, adjusted for transaction costs of $1.8 million. The loans were raised in USD and EUR to match revenue streams in USD and EUR. The remaining undrawn credit under the facilities were $74.1 million. Under the terms of the Facilities, the Group pledged the assets, including patents and other intellectual property, of our subsidiary, Olink Proteomics Inc.
On March 30, 2021, we repaid $65.6 million of outstanding loan facilities plus accrued interest of $1.9 million using the net proceeds from the offering and had no outstanding loan balances. As of December 31, 2023, we had $121.0 million in cash at bank and no outstanding loan balances or related pledged assets.
/81
Cash Flows
The table below summarizes our statement of cash flows for the periods presented:
| Year ended | ||||||||
| Amounts in thousands of USD | December 31, 2023 | December 31, 2022 | ||||||
| Cash flow used in operating activities | $ | (29,984) | (30,066) | |||||
| Cash flow used in investing activities | (22,434) | (8,713) | ||||||
| Cash flow (used in)/from financing activities | 92,496 | (2,884) | ||||||
| Net cash flow during the financial year | $ | 40,078 | (41,663) | |||||
Cash used in Operating Activities
Cash flow used in operating activities was $(30.0) million for the year ended December 31, 2023 and $(30.1) million for the year ended December 31, 2022. The negative cash flow from operating activities in 2023 is primarily explained by two components. One is loss before tax, adjusted with non-cash items, of $(20.6) million. The other is changes in working capital of $(14.0) million due to inventory buildup and higher accounts receivables both related to increased sales. Interest received and tax payment of $6.4 million and $(1.5) million respectively are two other components affecting cash flow in operating activities 2023. In 2022, the negative cash flow from operating activities is explained by net result before tax, adjusted for non-cash items, of $(4.6) million, a $(27.9) million change in net working capital and interest and tax payment of $(0.5) million and $1.3 million respectively.
Cash used in Investing Activities
Cash used in investing activities was $(22.4) million during the year ended December 31, 2023, representing a increase in cash used of $(13.7) million, or (157.5)%, compared to the year ended December 31, 2022. This increase resulted from increased investments in property, plant, and equipment of $(11.8) million, mainly due to new office in Sweden, and increased purchase of intangible assets of $(0.9) million in year ended December 31, 2023 compared to the year ended December 31, 2022.
Cash used in / provided by Financing Activities
Cash used in financing activities was $92.5 million during the year ended December 31, 2023, representing a increase in cash provided of $95.4 million. This increase primarily resulted from cash received from the issuance of new shares which amounted to $100.3 million the year ended December 31, 2023, partially offset by cash outflow from share issue costs of $(5.1) million. There was no repayment of interest-bearing debt and borrowings in the year ended December 31, 2023. Repayment of interest-bearing debt and borrowings amounted to $0.0 million the year ended December 31, 2022.
Operating and Capital Expenditure Requirements
Since our inception in 2016, we have incurred operating losses from time to time. Our net loss was $31.6 million during the year ended December 31, 2023 compared to a net loss of $12.9 million for the year ended December 31, 2022. We expect to incur significant expenses as well as operating losses during a period going forward as we continue our research and development efforts and expand our protein biomarker library. In addition, we plan to further expand our commercial team globally.
Although it is difficult to predict future liquidity requirements, we believe that our existing cash and cash equivalents as of December 31, 2023 will be sufficient to cover the planned funding need until the business is funded through a positive cash flow.
/82
Contractual Obligations
The following table discloses aggregate information about our material undiscounted contractual obligations and the periods in which payments are due as of December 31, 2023 and December 31, 2022. Future events could cause actual payments and timing of payments to differ from the contractual cash flows set forth below.
| As per December 31, 2023 | |||||||||||||||||
| Amounts in thousands of USD | Total | Less than 1 year | 1 - 3 years | 3 - 5 years | More than 5 years | ||||||||||||
| Lease obligations | $ | 32,721 | $ | 4,145 | $ | 11,285 | $ | 8,077 | $ | 9,214 | |||||||
| Accounts payable | 18,758 | 18,758 | — | — | — | ||||||||||||
| Royalties | 5,043 | 5,043 | — | — | — | ||||||||||||
| Salaries and wages | 9,530 | 9,530 | — | — | — | ||||||||||||
| Other liabilities | 938 | 938 | — | — | — | ||||||||||||
| As per December 31, 2022 | |||||||||||||||||
| Amounts in thousands of USD | Total | Less than 1 year | 1 - 3 years | 3 - 5 years | More than 5 years | ||||||||||||
Lease obligations (1) | $ | 35,528 | $ | 4,606 | $ | 8,255 | $ | 7,732 | $ | 14,935 | |||||||
| Accounts payable | 6,885 | 6,885 | — | — | — | ||||||||||||
| Royalties | 2,321 | 2,321 | — | — | — | ||||||||||||
| Salaries and wages | $ | 10,185 | $ | 10,185 | — | — | — | ||||||||||
| Other liabilities | 1,100 | 1,100 | — | — | — | ||||||||||||
1) Included lease agreements that the Group has entered into, but not yet commenced at December 31, 2022.
Loan facilities
Our loan facilities with Bridgepoint Credit and DNB AB (Publ) which were used as part of the financing of the Olink Acquisition (Facilities) and amounted to a total commitment under the facilities to $137.6 million was paid off in full during 2021. As of December 31, 2022 and 2023, we do not have outstanding loans.
Lease liabilities
Leases consist of real estate leases for our offices located in Uppsala, Umeå and Stockholm in Sweden, Waltham in Massachusetts, and Shanghai, China. Additionally, from time to time we enter into lease agreements for scientific equipment that contain a purchase option.
Advance invoiced customers
Represents cash receipts from customers which will be recognized as revenue upon completion of the related performance obligations.
Accounts Payable
Accounts payable represents amounts owed to vendors for purchases made in the ordinary course of business.
C.Research and Development, Patents and Licenses
For a description of our research and development programs and activities, see “Item 4.B. Information on the Company - Business Overview”. For a description of the amount spent during each of the last three fiscal years on company-sponsored research and development activities, as well as the four components of research and development expenses, see “Item 5. Operating and Financial Review and Prospects-A. Operating Results-Financial Operations Overview.”
/83
D.Trend Information
Other than as disclosed elsewhere in this annual report, we are not aware of any trend, uncertainty, demand, commitment or event that is reasonably likely to have a material effect on our net revenues and income from continuing operations, profitability, liquidity, capital resources, or would cause reported financial information not necessarily to be indicative of future operation results or financial condition.
During the year ended December 31, 2023 and December 31, 2022, we did not have any off-balance sheet arrangements.
E.Critical Accounting Estimates
See Note 3 to our consolidated financial statements for a summary of our material accounting policy information, estimates and judgments.
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
A.Directors and Senior Management
The following table sets forth the name and position of each of our executive officers and directors, as well as their respective ages as of March 15, 2024.
| Name | Age | Position(s) | ||||||
| Executive Officers: | ||||||||
| Jon Heimer | 56 | Chief Executive Officer and Director | ||||||
| Oskar Hjelm | 39 | Chief Financial Officer | ||||||
| Rickard El Tarzi | 37 | Chief Strategy and Product Officer | ||||||
| Maria Liminga Björk, PhD | 55 | Chief R&D Officer | ||||||
| Carl Raimond | 53 | President | ||||||
| Anna Marsell | 45 | Chief Operating Officer | ||||||
| Bruno Rossi | 51 | Chief Commercial Officer | ||||||
| Elias Berglund | 46 | Chief People Officer | ||||||
| Linda Ramirez-Eaves, Esq. | 52 | General Counsel | ||||||
| Directors: | ||||||||
| Jon Hindar | 67 | Chairman of the Board of Directors | ||||||
| Solange Bullukian | 59 | Director | ||||||
| Johan Lund, PhD | 66 | Director | ||||||
| Mary Reumuth | 48 | Director | ||||||
| Nicolas Roelofs, PhD | 66 | Director | ||||||
| Gregory J. Moore | 59 | Director | ||||||
| Tommi Unkuri | 43 | Director | ||||||
| Robert Scheuren | 62 | Director | ||||||
The following is a brief summary of the business experience of each of the individuals above:
Executive Officers
Jon Heimer has served as the chairman of our subsidiary, Olink Proteomics AB since 2014 and Chief Executive Officer of Olink Proteomics AB since January 2016, and has served as a member of our Board of Directors since December 2020. Prior to joining us, from April 2011 until December 2015, Mr. Heimer was a partner at Nexttobe AB, a family office/investment company focused on the Swedish biotechnology industry. Mr. Heimer has served as chairman of the board of directors of Q-linea AB, and for multiple privately-held biotechnology companies, including Bioimics AB and Lumina Adhesives AB. Mr. Heimer is a serial entrepreneur, was one of the key persons in successful Q-Med starting off in the 1990s and has spent a large part of his professional career working from the United States in various investments and growth companies within the biotech space.
/84
Oskar Hjelm has served as our Chief Financial Officer since March 2020. Prior to joining us, from September 2017 until February 2020, Mr. Hjelm worked at Alvarez & Marsal Sweden AB within their Transaction Advisory Group providing support to European and Nordic private equity funds. From August 2016 until August 2017, Mr. Hjelm was a director at KPMG AB. From January 2016 until August 2016, Mr. Hjelm was an investment controller at Nordic Capital. From July 2008 until December 2015, Mr. Hjelm held various roles at KPMG AB and KPMG LLP (United Kingdom). Mr. Hjelm received his Master of Science in business and economics from Linköpings University.
Rickard El Tarzi has served as our Chief Strategy Officer since February 2020 and served as a member of our Board of Directors from March 2019 to February 2020. Prior to joining us, from January 2017 until February 2020, Mr. El Tarzi served as an investment director on the investment team of Summa Equity AB. From April 2012 until April 2016, Mr. El Tarzi worked at McKinsey & Company advising investor and corporate clients across Europe and the United States on strategy and mergers and acquisitions. Mr. El Tarzi received his Bachelor of Science in logistics and transport management and his Master of Science in management from University of Gothenburg School of Business, Economics, and Law.
Carl Raimond has served as our President since March 2023, and previously served as our Chief Commercial Officer from October 2020 to March 2023 and as our Senior Vice President of Sales from August 2020 until October 2020. Prior to joining us, from January 2015 until February 2020, Mr. Raimond served in various executive commercial leadership roles at PerkinElmer, Inc. including Vice President and General Manager of Americas Sales and Service and Global Vice President and General Manager of Sales and Service for the Discovery and Analytical Solutions Division. From June 2010 until January 2015, Mr. Raimond served as the Vice President and General Manager of the Americas Life Science Sales & Field Operations of Agilent Technologies, Inc. Mr. Raimond received his Bachelor of Arts in zoology from State University of New York College at Oswego, and his Master of Science in biology from State University of New York College at Brockport.
Anna Marsell has served as our Chief Operating Officer since November 29, 2022. Prior to joining us, Ms. Marsell worked at Galderma from May 2012 to November 2022 in several different roles including Global Brand Manager, Global Strategic Marketing in Uppsala Sweden, Council Management and decision report, based in Switzerland and working directly with the CEO of Nestlé and since May 2019 as General Manager/Head of Nordics in Uppsala. Prior to that she was based in Boston working in Product Management for St. Jude Medical from April 2009 to May 2012. She started out her career at Radi Medical Systems in January 2005 working as a Project Manager in Uppsala until April 2008 when she moved to Boston working in Product Management until April 2009. Ms Marsell has a M.Sc in Bio Tech Engineering from Uppsala University from 1997 to 2004 and she also worked as a Research Engineer at Uppsala University from April 2004 to December 2004.
Bruno Rossi has served as our Chief Commercial Officer since March 2023. He brings more than 25 years of global experience in the life sciences industry, including the laboratory, pharmaceutical, and diagnostic market segments; and with senior strategic roles in product management, sales and marketing, and R&D. Before joining Olink, Bruno held multiple leadership positions at Leica Microsystems (a Danaher company) most recently serving as Vice President Global Business Transformation. Previous to Leica Microsystems, Bruno spent 22 years at Millipore (now part of Merck KGaA) holding various senior positions in sales, marketing, and product management. Bruno holds a Master’s degree in Organic Chemistry from École Nationale Supérieure de Chimie de Clermont-Ferrand (ENSCCF).
Elias Berglund has served as our Chief People Officer since May 2023, and brings more than 15 years of human resources experience at global and growing companies across different industries. Prior to joining Olink, from August 2020 until April 2023, Mr. Berglund worked at Universum Communications AB as Global Chief HR Officer. Prior to that he served seven years in various positions, mainly as Chief HR Officer at Tre AB; with additional experience from Klarna and SF Bio AB. Mr. Berglund studied behavioral science in Stockholm.
Linda Ramirez-Eaves, Esq. has served as our General Counsel since February 2019. Prior to joining us, from December 2018 to February 2019, Ms. Ramirez-Eaves served as Senior Corporate Counsel for Seagate Technologies, and from September 2015 until December 2018, Ms. Ramirez-Eaves served as Senior Counsel of SomaLogic, Inc. From December 2014 until September 2015, Ms. Ramirez-Eaves served as Senior Legal Counsel at Ciber Global, LLC. Ms. Ramirez-Eaves received her Bachelor of Science in Journalism and Mass Communications from the University of Colorado at Boulder, and her Juris Doctorate from the University of Colorado at Boulder School of Law. Ms. Ramirez-Eaves has been a Certified Information Privacy Professional/Europe since 2018.
Maria Liminga Björk (PhD) has served as our Chief R&D Officer since January 2024. She joined Olink Proteomics as VP President R&D in March 2023 bringing more than 20 years of expertise from the Life Science industry. Prior to Olink, 2020-2023, she served as Director of BioProcess R&D at Cytiva, a Danaher Corp subsidiary. In this role Maria led an organization responsible for development of novel purification solutions for the Biopharma industry. From 2016- 2020 Maria directed the Resin and Ligand Department at GE Healthcare. From 2013-2016 Maria served as Head of the Global project office at GE Healthcare, managing a project portfolio covering cell culture media, cell culture bioreactors, filtration and chromatography systems and consumables. Maria commenced her career at Amersham Biosciences in 2000 as
/85
Scientist and thereafter Senior Scientist in Proteomics R&D. She has a Master’s in Pharmaceutical Biosciences and a PhD in Biochemical Pharmacology from the Uppsala University.
Directors
Jon Hindar has served as chairman of our Board of Directors since January 2021. Mr. Hindar has served as a Principal of Summa Equity AB since January 2017. From 2015 until 2017, Mr. Hindar served as chairman of the board of directors of Argentum Fondsinvesteringer AS, Hav Line AS and LGJ Invest AS. From March 2012 until June 2016, Mr. Hindar served as Chief Executive Officer of Cermaq Group AS. Mr. Hindar has served as chairman of the board of directors of Arendals Fossekompani ASA since June 2020, and also serves on the boards of multiple privately-held companies, including Milarex AS, Klaveness Marine Holding AS, LGJ Invest AS, HyTest Group, Argentum Fondsinvesteringer AS and Nofitech AS. Mr. Hindar received his Master of Science and Engineering in chemistry from the Norwegian University of Science and Technology, and completed the Programme for Executive Development at IMD, Lausanne. We believe Mr. Hindar is qualified to serve on our Board of Directors because of his scientific knowledge, extensive business and operations experience, including in leadership roles, and his experience working with companies in similar technologies and markets.
Solange Bullukian has served as a member of our Board of Directors since January 2021. Ms. Bullukian is a strategic executive finance and accounting leader with extensive Fortune 500 and startup experience, including in the life sciences, technology, and computing industries. Ms. Bullukian is the Managing Principal of Scale2Growth which she founded in November 2017, supporting companies through periods of rapid expansion. Ms. Bullukian served as the Chief Financial Officer of Twist Bioscience Corporation. Previously, Ms. Bullukian has served as Chief Accounting Officer and prior to that as Chief Financial Officer of the Life Sciences Group at Agilent Technologies Inc. Ms. Bullukian held a variety of finance and accounting positions at both Agilent Technologies and Hewlett-Packard. Ms. Bullukian is an Independent Director and Audit Committee Chair at Lumicks and Inari Agriculture. Ms. Bullukian received her Master of Science in Management from the HEC (Ecole des Hautes Etudes Commerciales) School of Management in Paris, France. We believe Ms. Bullukian is qualified to serve on our Board of Directors because of her experience, qualifications, attributes and skills, including her experience in the emerging growth and life sciences markets and her service as a director of other companies.
Johan Lund, PhD has served as a member of our Board of Directors since December 2020. He has served as the co- founder and Chief Executive Officer of KyNexis Medicine Development AB since August 2018. Since June 2018, Dr. Lund has also served as a consultant for MBS Pharma, which he founded. Prior to that, from March 2016 until May 2017, Dr. Lund served as Vice President and Head of the Immunology and Inflammation Therapeutic Center of Excellence of Celgene Corporation. From April 2015 until March 2016, Dr. Lund was Managing Partner at J. Lund and Associates, LLC, and from May 2015 until March 2016, Dr. Lund was a Senior Advisor for the Karolinska Institutet, advising on innovation and business creation as part of the European Institute for Innovation and Technology (EIT) Health Consortium. From August 2012 until March 2015, Dr. Lund served as Senior Vice President and Chief Scientific Officer of the Immunoscience Research Unit of Pfizer Inc. Dr. Lund has served as chairman of the board of directors for Aqilion AB since June 2018, and is a member of the board of directors of several privately-held companies, including Genagon Therapeutics AB and NEOGAP AB (formerly Tcer AB). Dr. Lund received his Med.Kand. degree and his Doctor of Medical Science degree from Karolinska Institutet. Dr. Lund also holds a diploma in Managing Medical Product Innovation from the Scandinavian International Management Institute in Copenhagen. We believe Dr. Lund is qualified to serve on our Board of Directors because of his extensive medical and scientific knowledge and his extensive operating experience in the biotechnology industry.
Mary Reumuth has served as a member of our Board of Directors since April 2022. She is currently the CFO of Kala Pharmaceuticals, a publicly traded biopharmaceutical company focusing on advancing the treatment of eye diseases. Ms. Reumuth acted as an independent financial consultant from November 2012 to January 2014, and served as Corporate Controller for Enobia Pharma Corp., a biopharmaceutical company acquired by Alexion Pharmaceuticals, Inc., from May 2011 to June 2012. She previously served as Director of Finance at Verenium Corporation, a biotechnology company, from December 2007 to March 2011. From 2001 to 2007, Ms. Reumuth held a variety of finance and accounting positions at Genzyme Corporation, and ILEX Oncology, Inc. Ms. Reumuth has served an auditor at Ernst & Young LLP. She earned her bachelor's degree in Business Administration from Texas A&M University—Corpus Christi, and is a Certified Public Accountant. We believe Ms. Reumuth is qualified to serve on our Board of Directors because of her operating experience in leading financial functions in the biopharmaceutical industry.
Nicolas Roelofs, PhD has served as a member of our Board of Directors since December 2020. Dr. Roelofs has served as a Principal of Summa Equity AB since July 2019. Dr. Roelofs has also served as Industrial Advisor of Nordic Capital since 2014. Dr. Roelofs serves as chairman of the board of directors of multiple privately-held companies, including Sengenics Corporation Pte Ltd., One BioMed Pte Ltd., ScaleBio Ltd., and Boreal Genomics Inc. Dr. Roelofs also serves as a member of the board of directors of multiple privately-held companies, including HyTest Ltd., The Binding Site Group Ltd., InSilixa, Inc., and LGC Group. He also serves as an advisory board member of 908 Devices Inc. Dr. Roelofs previously served as the President of the Life Sciences Group at Agilent Technologies, Group Operations Officer for the Life Sciences Division of Bio-Rad Inc., and Chief Operating Officer of Stratagene Inc. Dr. Roelofs received his Bachelor of
/86
Science in chemistry, biology, and German from Simpson College, his Master of Science in organic chemistry from Iowa State University, and his doctorate in organic chemistry from University of Nevada, Reno. We believe that Dr. Roelofs is qualified to serve on our Board of Directors because of his experience, qualifications, attributes and skills, including his scientific knowledge, extensive experience in the life sciences and healthcare markets, and his service as a director of other companies.
Dr. Gregory James Moore has served as a member of our Board of Directors since April 2023. Dr. Moore served as Corporate Vice President for Microsoft from 2019-2023, most recently leading global health and life sciences, and prior leading health technology and alliances. Before joining Microsoft, Dr. Moore served as Vice President, Google and was founder of Google Cloud Healthcare and Life Sciences since 2016. Dr. Moore is board certified in Diagnostic Radiology, Neuroradiology, and Clinical Informatics. Prior to his executive leadership roles at Microsoft and Google, Dr. Moore served as the chief emerging technology and informatics officer at Geisinger Health System, where he was also Director of the Institute of Advanced Application, Interim Chair of System Radiology and a practicing neuroradiologist. His prior academic and clinical appointments include Stanford University School of Medicine, Penn State University College of Medicine, and Wayne State University School of Medicine. He currently serves as an independent director on the board of DaVita and is a member of their Nominating and Governance, and Quality and Compliance committees. Dr. Moore also served as an independent director on the board of Hillrom including on their Compensation and Management Development and Merger and Acquisition committees until its acquisition by Baxter. Dr. Moore received his Bachelor of Science in Combined Sciences (Physics/Biology) from North Park College, his Master of Science in Nuclear Engineering from Massachusetts Institute of Technology (MIT), his doctorate in Radiological Sciences from MIT, his Doctor of Medicine degree from Wayne State University School of Medicine. We believe Dr. Gregory Moore is qualified to serve on our Board of Directors because of his experience, qualifications, attributes and skills, including his scientific knowledge, extensive experience in the healthcare and technology markets.
Tommi Unkuri has served as a member of our Board of Directors since March 2019. Mr. Unkuri has served as a Partner of Summa Equity AB since May 2016. From November 2015 until May 2016, Mr. Unkuri was a Partner at Fidelio Capital AB, and from April 2007 until December 2015, Mr. Unkuri worked with investments at Nordic Capital AB. Mr. Unkuri currently serves as a member of the board of directors of multiple privately- held companies, including Sengenics Corporation Pte Ltd., LOGEX Group and HyTest Ltd. Mr. Unkuri received his Master of Science from the Stockholm School of Economics. We believe Mr. Unkuri is qualified to serve on our Board of Directors because of his experience, qualifications, attributes and skills, including his financial expertise, investment experience, and his current and previous service as a director of other companies in the healthcare industry.
Robert Schueren has served as a member of our Board of Directors since April 2022. He currently serves as COO of Natera, a publicly traded company focused on women’s health, oncology, and organ health diagnostics. Prior to Natera, he was CEO of IntegenX Inc. until its acquisition by Thermo Fisher Scientific. Additional executive leadership roles that Mr. Schueren held include GM of Genomics at Agilent Technologies, Global Head of Clinical Biomarkers and Operations, and Deputy Global Head of Molecular Medicine Labs for Genentech, Inc. He formerly held leadership and commercial roles at Arcturus Bioscience, Accumetrics, Biosite Diagnostics, Gen-Probe, and Abbott Labs. Mr. Schueren received a BS in Pharmacy from Temple University. We believe Mr. Schueren is qualified to serve on our Board of Directors because of his experience, qualifications, attributes and skills, including his scientific knowledge, extensive operating experience in the life sciences and healthcare markets.
/87
B.Compensation
Executive Officer and Non-Executive Director Compensation
Our Chief Executive Officer and non-executive directors received the following compensation, accrued or paid, for the year ended December 31, 2023 (in USD):
| Base Pay | Variable/Bonus Pay | Pension Cost | Share-based Compensation 2 | Total | |||||||||||||
| Name and Title or Position | (US$) | (US$) | (US$) | (US$) | (US$) | ||||||||||||
| Chief Executive Officer and Director | |||||||||||||||||
Jon Heimer (1) | $ | 422,555 | $ | 208,459 | $ | 64,154 | $ | 828,437 | $ | 1,523,605 | |||||||
| Non-Executive Directors | |||||||||||||||||
| Jon Hindar | 130,000 | 128,453 | 258,453 | ||||||||||||||
| Solange Bullukian | 100,000 | 78,367 | 178,367 | ||||||||||||||
| Johan Lund, PhD | 90,000 | 78,367 | 168,367 | ||||||||||||||
| Mary Reumuth | 85,000 | 54,042 | 139,042 | ||||||||||||||
| Nicolas Roelofs, PhD | 70,000 | 78,367 | 148,367 | ||||||||||||||
| Gregory J. Moore | 59,500 | 30,103 | 89,603 | ||||||||||||||
| Tommi Unkuri | — | — | — | ||||||||||||||
| Robert Scheuren | 85,000 | 54,042 | 139,042 | ||||||||||||||
(1) Includes compensation for service as Chief Executive Officer; Mr. Heimer does not receive compensation for his service as a Director.
(2) Refers to RSUs granted to Mr. Heimer and options granted to all directors, where amounts indicated. Amounts represent the expense recognized in accordance with IFRS 2 in the income statement, based on the grant date fair value.
During and for the year ended December 31, 2023, the aggregate compensation accrued or paid to our other executive officers serving during the year as a group (nine individuals, including our former Chief People Officer who served through March, 2023) was base pay of US $2,166,578, variable/bonus pay of US $469,480, pension cost of US $343,333, and share based compensation of $2,065,633. Our executive officers also had amounts paid to provide healthcare benefits.
Mr. Heimer was granted stock options to purchase 17,045 shares in April 2023 with an exercise price of $22.79 per share and 38,182 RSUs. One-quarter of Mr. Heimer's options and RSUs vest annually on the first, second, third, and fourth anniversary after grant date, and the expiration date for each tranche of options is five years after the grant date.
For the non-executive directors, excluding Mr. Unkuri whom did not receive any compensation, each was granted stock options to purchase 6,909 shares in April 2023 with an exercise price of $22.79 per share. One-quarter of the options vest annually on the first, second, third, and fourth anniversary after grant date, and the expiration date for each tranche of options is five years after the grant date.
Our executive officers, including our Chief Executive Officer, participate in our performance based cash bonus incentive plan, which uses a balanced weighting of multiple performance measures and metrics to determine incentive payouts to our executive officers. The plan provides for annual cash incentive awards based on overall Company performance and individual performance and contribution. Our Board of Directors sets the performance objectives for the Company under the annual cash incentive plan.
For share-based compensation information for the year ended December 31, 2023 for our executive officers and non- executive directors, see "Amended and Restated 2021 Incentive Award Plan" below and "Note 21- Stock-based Compensation" in the Notes to the Consolidated Financial Statements contained herein.
Amended and Restated 2021 Incentive Award Plan
On March 16, 2021, our shareholders approved and made effective our 2021 Incentive Award Plan, which was subsequently amended and restated, and approved by our shareholders on April 7, 2022 (the Plan). On April 17, 2023, the 2021 Incentive Award Plan was amended solely to increase the number of shares covered therein by 980,000 shares as approved by the shareholders. The principal purpose of the Plan is to attract, retain and motivate selected employees, consultants and
/88
directors through the granting of share-based compensation awards and cash-based performance bonus awards. The material terms of the Plan are summarized below.
Under the Plan, a total of 2,660,303 Shares have been approved for issuance pursuant to a variety of stock-based compensation awards, including stock options, stock appreciation rights, or SARs, restricted stock unit awards, performance bonus awards, performance stock unit awards, dividend equivalents, other stock-based awards, and other cash-based awards; provided, however, that no more than 1,680,303 Shares may be issued upon the exercise of incentive stock options. “Shares” means, as determined by the administrator, (i) common shares or (ii) an equivalent number of American Depositary Shares or American Depositary Receipts, provided, however, it is understood that in order to facilitate the delivery and settlement of an award, an award may be settled by delivering warrants, entitling the holder to the immediate subscription of one common share against the (at the time) quota value of such common share, and which shall be immediately converted into common shares.
The following counting provisions are in effect for the shares available under the Plan:
•to the extent that an award terminates, expires or lapses for any reason or an award is settled in cash without the delivery of Shares, any Shares subject to the award at such time will be available for future grants under the Plan;
•to the extent Shares are tendered or withheld to satisfy the exercise price or tax withholding obligation with respect to any award under the Plan, such tendered or withheld Shares will be available for future grants under the Plan, provided it is permitted under applicable law;
•to the extent Shares subject to stock appreciation rights are not issued in connection with the settlement of stock appreciation rights on exercise thereof, such Shares will be available for future grants under the Plan;
•any Shares that are subject to awards that may only be settled in cash will not be counted against the Shares available for issuance under the Plan; and
•to the extent permitted by applicable law or any exchange rule, Shares issued in assumption of, or in substitution for, any outstanding awards of any entity acquired in any form of combination by us or any of our subsidiaries will not be counted against the Shares available for issuance under the Plan.
For share-based compensation information for the year ended December 31, 2023 for our executive officers and non- executive directors, see "Note 21- Stock-based Compensation" in the Notes to the Consolidated Financial Statements contained herein.
C.Board practice
Introduction
Our Board of Directors performs its duties in accordance with the Rules of Procedure for the Board of Directors of Olink Holding AB (publ) and the Swedish Companies Act. The Rules of Procedure are reviewed and adopted by the Board of Directors annually. Our Board of Directors, including the Chairman, is elected by our shareholders at the Annual General Meeting to serve until the end of the next Annual General Meeting, with the possibility of re-election, or until their earlier removal or resignation. The majority of our Board members are considered to be independent under the independence standards of Nasdaq.
Corporate governance
We are a “foreign private issuer,” as defined by the SEC. As a result, in accordance with Nasdaq listing requirements, we may rely on home country governance requirements and certain exemptions thereunder rather than complying with Nasdaq corporate governance standards. While we voluntarily follow most Nasdaq corporate governance rules, we may choose to take advantage of the following limited exemptions:
•exemption from filing quarterly reports on Form 10-Q containing unaudited financial and other specified information or current reports on Form 8-K upon the occurrence of specified significant events;
•exemption from Section 16 rules requiring insiders to file public reports of their securities ownership and trading activities and providing for liability for insiders who profit from trades in a short period of time;
/89
•exemption from the Nasdaq requirement necessitating disclosure of any waivers of the Code of Conduct for directors and executive officers;
•exemption from the requirement to obtain shareholder approval for certain issuances of securities, including shareholder approval of share option plans;
•exemption from the requirement that our Audit Committee have review and oversight responsibilities over all “related party transactions,” as defined in Item 7.B of Form 20-F;
•exemption from the requirement that our Board of Directors have a compensation committee that is composed entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities; and
•exemption from the requirement to have independent director oversight of director nominations.
Furthermore, Nasdaq Rule 5615(a)(3) provides that a foreign private issuer may rely on home country corporate governance practices in lieu of certain of the rules in the Nasdaq Rule 5600 Series and Rule 5250(d). We follow Swedish corporate governance practices in lieu of Nasdaq corporate governance requirements as follows:
•We do not follow Nasdaq Rule 5620(c) regarding quorum requirements applicable to meetings of shareholders. Such quorum requirements are not required under Swedish law. The Swedish Companies Act (SFS 2005:551) and our Articles of Association provide alternative quorum requirements that are generally applicable to meetings of shareholders.
•We do not follow Nasdaq Rule 5605(b)(2), which requires that independent directors regularly meet in executive sessions where only independent directors are present. Our independent directors may choose to meet in executive sessions at their discretion.
•We do not follow Nasdaq Rule 5605(d) regarding the composition of the Remuneration Committee.
•We do not follow Nasdaq Rule 5605(e) regarding the composition of the Nominating Committee
Although we may rely on certain home country corporate governance practices, we must comply with Nasdaq’s Notification of Noncompliance requirement (Nasdaq Rule 5625) and the Voting Rights requirement (Nasdaq Rule 5640). Further, we must have an audit committee that satisfies Nasdaq Rule 5605(c)(3), which addresses Audit Committee responsibilities and authority and requires that the Audit Committee consist of members who meet the independence requirements of Nasdaq Rule 5605(c)(2)(A)(ii). Because we are a foreign private issuer, our directors and executive officers are not subject to short- swing profit and insider trading reporting obligations under Section 16 of the Exchange Act. They are, however, subject to the obligations to report changes in securities ownership under Section 13 of the Exchange Act and related SEC rules.
We intend to continue to take all actions necessary for us to maintain compliance as a foreign private issuer under the applicable corporate governance requirements of the Sarbanes-Oxley Act, the rules adopted by the SEC and Nasdaq listing rules.
Accordingly, our shareholders do not and in the future will not have the same protections afforded to shareholders of companies that are subject to all of the corporate governance requirements of Nasdaq. For an overview of our corporate governance principles, see the section titled “Item 10. Additional Information - Memorandum and Articles of Association - Differences in Corporate Law.
In addition to being a foreign private issuer, we are also a “controlled company” within the meaning of the corporate governance rules of Nasdaq, as Knilo InvestCo AS, which is owned by several funds controlled by Summa Equity AB, controls a majority of the voting power of our outstanding common shares. As a “controlled company,” certain exemptions under the Nasdaq listing standards free us from the obligation to comply with certain Nasdaq corporate governance requirements, including the requirements:
•that a majority of our Board of Directors consist of “independent directors,” as defined under Nasdaq rules;
•that our Board of Directors have a Remuneration Committee that is comprised entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities and
•that our Board of Directors have a Nominating and Corporate Governance Committee that is comprised entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities.
/90
Accordingly, stockholders do not and in the future will not have the same protections afforded to stockholders of companies that are subject to all of the corporate governance rules of Nasdaq. These exemptions do not modify the independence requirements for our Audit Committee.
Composition of Our Board of Directors
Our Board of Directors is comprised of nine members. Under the rules and regulations of Nasdaq, a director will qualify as “independent” if our Board of Directors affirmatively determines that he or she has no material relationship with us (either directly or as a partner, shareholder or officer of an organization that has a relationship with us). Our Board of Directors has determined that, of our nine directors, no director, other than Jon Heimer and Tommi Unkuri, has a relationship that would interfere with the exercise of independent judgment in carrying out his or her responsibilities as a director and that each of these directors is “independent” as that term is defined under Nasdaq rules.
Our Board of Directors performs its duties in accordance with the Rules of Procedure for the Board of Directors of Olink Holding AB (publ). The Rules of Procedure are reviewed and adopted by the Board of Directors annually. Our Board of Directors, including the chairman, is elected by our shareholders at the annual shareholders’ meeting up until the end of the next annual shareholders’ meeting, with the possibility of re-election.
Committees of Our Board of Directors Audit Committee
Our Audit Committee consists of Solange Bullukian, Mary Reumuth, and Robert Scheuren, who are responsible for overseeing our accounting and financial reporting processes. Solange Bullukian serves as chair of the Audit Committee. The Audit Committee consists exclusively of members of our Board who are financially literate, and Solange Bullukian is considered an “audit committee financial expert” as defined by applicable SEC rules and has the requisite financial sophistication as defined under the applicable Nasdaq rules and regulations. The Audit Committee is governed by a charter that complies with Nasdaq rules.
The Audit Committee’s responsibilities include, among others:
•monitoring our financial reporting;
•monitoring the efficiency of our internal controls, internal audit activities and risk management;
•keeping informed of the auditing of the annual report and the consolidated accounts;
•reviewing and monitoring the impartiality and independence of our auditors and paying close attention to whether our auditors are providing other services besides audit services for us; and
•assisting in the preparation of proposals for our shareholders’ meeting’s election of auditors; and
•monitoring sustainability reporting and overseeing sustainability topics, including business risks and opportunities,
Remuneration Committee
Our Remuneration Committee consists of Gregory J. Moore, Johan Lund, Tommi Unkuri, and Jon Hindar. Johan Lund serves as chairman of the Remuneration Committee. The Remuneration Committee’s responsibilities include, among others:
•identifying, reviewing and proposing policies relevant to the compensation and benefits of our executive officers;
•evaluating each executive officer’s performance in light of such policies and reporting to the Board;
•overseeing and administering our employee share option scheme or equity incentive plans in operation from time to time; and
•review and assess the Company’s development plans for active talent. Report assessment and recommendations to the Board.
/91
D.Employees
As of December 31, 2023, we had 707 employees, including a recently increased commercial team of 245 individuals and an Research and Development (R&D) team of 82 individuals. The majority of our employees operate out of our Uppsala, Sweden headquarters. We also have secondary headquarters in Waltham, Massachusetts and a growing footprint across Singapore, China and Japan. We plan to continue investing in the development of our employees and promoting our culture of customer service and support through innovation, quality, rigor and transparency, as well as fostering our shared vision to enable understanding of real-time human biology. As of December 31, 2022 we had 582 employees including commercial team of 208 individuals and an R&D team of 70 individuals. As of December 31, 2021 we had 416 employees whereof 150 individuals in the Commercial team and 89 employees in our R&D team.
| Department | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||
Commercial | 245 | 208 | 150 | ||||||||
Research and Development (R&D) | 82 | 70 | 89 | ||||||||
Other | 380 | 304 | 177 | ||||||||
Total | 707 | 582 | 416 | ||||||||
E.Share Ownership
The share ownership information with respect to executive officers and the Board of Directors is presented in Item 6(B) above and Item 7 below.
F.Disclosure of a registrant’s action to recover erroneously awarded compensation
Not applicable.
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A.Major Shareholders
The following table sets forth information with respect to the beneficial ownership of our common shares as of February 5, 2024 for:
•each beneficial owner of 5% or more of our outstanding common shares;
•each of our directors and executive officers; and
•all of our directors and executive officers as a group.
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include common shares that can be acquired within 60 days of February 5, 2024. Percentage ownership calculations for shares beneficially owned are based on 124,342,715 common shares outstanding as of February 5, 2024 and include common shares held in the form of ADSs.
Except as otherwise indicated, shares reflected in the table are common shares and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
/92
Except as otherwise indicated in the table below, addresses of the directors, executive officers and named beneficial owners are c/o Olink Holding AB (publ), Salagatan 16F, SE-753 30, Uppsala, Sweden.
| Shares beneficially owned at 31 December 2023 | Beneficial shares vesting in 60 days | Shares beneficially owned at 5 February 2024 including shares issued and vesting within 60 days | ||||||||||||||||||
| Name of Beneficial Owner | Shares | % | RSUs | Options | Shares | % | ||||||||||||||
| 5% or Greater Shareholders: | ||||||||||||||||||||
Summa Equity AB(1) | 77,284,718 | 62.15 | % | — | — | 77,284,718 | 62.04 | % | ||||||||||||
| Fidelity Investments Canada ULC | 8,127,000 | 6.54 | % | — | — | 8,127,000 | 6.52 | % | ||||||||||||
| Executive Officers and Directors: | ||||||||||||||||||||
Jon Heimer(2) | 3,103,790 | 2.50 | % | 26,961 | 30,186 | 3,160,937 | 2.54 | % | ||||||||||||
| Oskar Hjelm | 220,740 | * | 10,307 | 6,097 | 237,144 | * | ||||||||||||||
Rickard El Tarzi(3) | 343,868 | * | 6,482 | 3,431 | 353,781 | * | ||||||||||||||
| Maria Liminga Björk, PhD | — | * | 1,643 | — | 1,891 | * | ||||||||||||||
| Carl Raimond | 263,555 | * | 14,922 | 7,239 | 285,716 | * | ||||||||||||||
| Linda Ramirez- Eaves, Esq | 23,489 | * | 9,663 | 4,669 | 37,821 | * | ||||||||||||||
Jon Hindar(4) | 208,954 | * | — | 30,860 | 239,814 | * | ||||||||||||||
| Solange Bullukian | 19,858 | * | — | 12,829 | 32,687 | * | ||||||||||||||
| Johan Lund, PhD | 60,703 | * | — | 12,829 | 73,532 | * | ||||||||||||||
| Nicolas Roelofs, PhD | 152,892 | * | — | 12,829 | 165,721 | * | ||||||||||||||
| Gregory J. Moore | — | — | — | 1,728 | 1,728 | * | ||||||||||||||
| Mary Reumuth | 2,344 | * | — | 4,072 | 6,416 | * | ||||||||||||||
| Robert Schueren | 2,344 | * | — | 4,072 | 6,416 | * | ||||||||||||||
| Tommi Unkuri | — | — | — | — | — | — | ||||||||||||||
| Bruno Rossi | 1,931 | * | 7,625 | 831 | 10,387 | * | ||||||||||||||
Erika Assarsson(5) | 275,323 | * | 4,500 | 1,545 | 281,368 | * | ||||||||||||||
| Torbjörn Wärnheim | 558 | * | 1,123 | — | 1,681 | * | ||||||||||||||
Andrea Ballagi(6) | 724,072 | * | 4,252 | — | 728,324 | * | ||||||||||||||
Bill Campbell(7) | 147,166 | * | 5,448 | — | 152,614 | * | ||||||||||||||
| Anna Marsell | 1,953 | * | 6,283 | 524 | 8,760 | * | ||||||||||||||
| Elias Berglund | — | * | 1,870 | — | 1,870 | * | ||||||||||||||
*Represents beneficial ownership of less than one percent.
1.Summa Equity AB, indirectly through intermediary funds and coinvestment entities, is the sole shareholder of Knilo InvestCo AS. Summa Equity AB has also been designated as the sole manager of such intermediary funds and co- investment entities. Summa Equity AB is authorized by the Swedish Financial Supervision Authority (the SFSA) to conduct business under the Alternative Investment Fund Managers Directive (2011/61/EU) (as enacted in Sweden) and is thereby under the supervision of the SFSA. The voting and dispositive decisions of Summa Equity AB are made by its board of directors, the members of which are Reynir Indahl, Eva Broms, Martin Skancke and Mirja Lehmler-Brown. The address of each of Summa Equity AB, the intermediary funds and coinvestment entities and the individuals mentioned herein is c/o Summa Equity AB, Birger Jarlsgatan 27, 111 45 Stockholm. We are currently controlled by Summa Equity AB. In connection with the Offer and Purchase Agreement with Thermo Fisher described in Items 4.A and 8.B of this report, Summa Equity AB and additional shareholders and management entered into support agreements agreeing to tender into the tender offer. The Tender and Support Agreement by and between Thermo Fisher Scientific Inc. and certain shareholders of Olink Holding AB (publ), dated October 17, 2023, is filed as Exhibit 4.18 to this report.
2.Consists of common shares held indirectly (through an endowment insurance) by Jon Heimer Invest AB. Voting and investment decisions with respect to common shares held by Jon Heimer Invest AB are made by Jon Heimer.
/93
3.Consists of common shares held indirectly (through an endowment insurance) by Heistbaron Togwaggle AB. Voting and investment decisions with respect to common shares held by Heistbaron Togwaggle AB are made by Rickard El Tarzi.
4.Consists of common shares held by Petrus Holding AS. Voting and investment decisions with respect to common shares held by Petrus Holding AS are made by Jon Hindar.
5.The Group initiated a public offering January 18, 2023. For more information see item 5.B Liquidity and Capital Resources.
6.Consists of ADSs held by Dalama AB. Voting and investment decisions with respect to common shares held by Dalama AB are made by Andrea Ballagi. Ms. Ballagi is a current employee of the company.
7.Consists of common shares and ADSs held by Mr. Campbell, a current employee of the Company.
B.Related party transactions
Within this section, we have calculated the U.S. dollar amounts using the historical exchange rate as of the date of each transaction. Other than compensation arrangements described in “Management” elsewhere in this Annual Report, since January 1, 2023, we have not entered into any transactions with our executive officers, directors or holders of more than 5% of our share capital, including their affiliates, which we refer to as our related parties.
Transactions with related parties prior to January 1, 2023 and still in place during 2023 included the following:
Agreements with Our Executive Officers and Directors
We have entered into employment agreements with certain of our executive officers. These agreements contain customary provisions and representations, including confidentiality, non-competition, non-solicitation and inventions assignment undertakings by the executive officers and non-executive directors. The enforceability of the non-competition provisions may be limited under applicable law.
Agreements with Shareholders
In connection with our initial public offering in March 2021, we entered into a Registration Rights Agreement and Amended and Restated Shareholders Agreement with certain holders of our common shares, which provide for certain rights, including rights of first refusal and co-sale and drag along rights and registration rights. See ITEM 10. ADDITIONAL INFORMATION for additional information.
Related Party Transactions Policy
In connection with our initial public offering, we adopted a Related Party Transaction Policy requiring that all related party transactions required to be disclosed by a foreign private issuer pursuant to the Exchange Act be approved by the audit committee or another independent body of our board of directors.
C.Interests of Experts and Counsel
Not applicable.
ITEM 8. FINANCIAL INFORMATION
A.Consolidated Statements and Other Financial Information
Refer to Item 18. Financial Statements herein for our Consolidated Financial Statements and report of our independent registered public accounting firm, Ernst & Young AB, Stockholm, Sweden (PCAOB No. 1433).
A.1Legal Proceedings
From time to time, we may be involved in various claims and legal proceedings relating to claims arising out of our operations. We are not involved in any legal or arbitration proceedings, including those relating to bankruptcy, receivership or similar proceedings and those involving any third party, which may have, or have had in the recent past, significant
/94
effects on our financial position or profitability. The company is not involved in any governmental proceedings pending or known by us to be contemplated, which may have, or have had in the recent past, significant effects on our financial position or profitability.
A.2Dividend Distribution Policy
We have never paid cash dividends to our shareholders. We intend to retain future earnings for use in our business and do not anticipate paying cash dividends on our ordinary shares in the foreseeable future. Any future dividend policy will be determined by the Board of Directors and will be based upon various factors, including our results of operations, financial condition, current and anticipated cash needs, future prospects, contractual restrictions and other factors as the Board of Directors may deem relevant. There is no assurance that dividends will ever be paid. See "Risk Factors" and “Special Note Regarding Forward Looking Statements” contained herein.
B.Significant Changes
On October 17, 2023, the Company entered into the Purchase Agreement with Thermo Fisher.
Pursuant to the terms of the Purchase Agreement, on October 31, 2023, Buyer (i) commenced the Offer, a cash tender offer to purchase all of the outstanding Common Shares of the Company and all of the Company’s outstanding American Depositary Shares, each of which represents one Common Share (collectively, the “ADSs,” and, together with the Common Shares, the “Offer Securities”), in exchange for $26.00 per Common Share, representing $26.00 per ADS, in cash, without interest (such amount or any higher amount per Common Share and ADS paid pursuant to the Offer in accordance with the Purchase Agreement, the “Offer Consideration”) and (ii) with respect to the Company Stock Options and Company RSUs (as such terms are defined in the Purchase Agreement) outstanding immediately prior to the Offer closing, implemented arrangements to (A) cash out Company Stock Options vested but unexercised as of immediately prior to the Offer closing in exchange for an amount in cash equal to the product of (1) the number of Common Shares subject to the vested portion of the underlying award and (2) the excess, if any, of the Offer Consideration over the applicable award exercise price and (B) otherwise convert such unvested Company Stock Options (if the exercise price is less than the Offer Consideration) and Company RSUs into restricted cash awards based on the Offer Consideration, with each converted award (1) vesting on the same terms and conditions applicable to the original awards after the Offer closing and (2) upon the terms and subject to the conditions set forth in the Purchase Agreement. Company Stock Options outstanding as of immediately prior to the Offer closing with an exercise price equal to or greater than the Offer Consideration will be cancelled for no consideration.
The Offer was set to initially expire one minute after 11:59 p.m. (New York City time) on the day that was 20 business days following the commencement of the Offer, and was extended in accordance with the terms of the Purchase Agreement, including as required by the applicable rules and regulations of the SEC to 5:00 p.m., New York City Time on April 30, 2024 (such date, the “Expiration Time”).
The obligation of Buyer to consummate the Offer is subject to customary conditions, including, among others, that immediately prior to the expiration of the Offer, (i) there have been validly tendered in accordance with the terms of the Offer, and not properly withdrawn, a number of Offer Securities (excluding Offer Securities tendered pursuant to guaranteed delivery procedures that have not yet been delivered in settlement or satisfaction of such guarantee prior to the Expiration Time) that, together with the Offer Securities then owned by Buyer or its affiliates and the Offer Securities that will be transferred to Buyer pursuant to the Tender and Support Agreement (as defined below) at the Offer closing, represents at least one Common Share more than 90% of the issued and outstanding Common Shares (excluding any Common Shares held in treasury by Olink or owned by any of Olink’s subsidiaries) immediately prior to the Expiration Time (the “Minimum Tender Condition”), provided that Buyer has the right to waive or decrease the Minimum Tender Condition to a percentage that is no lower than 51% of the issued and outstanding Common Shares (excluding any Common Shares held in treasury by Olink or owned by any of Olink’s subsidiaries); and (ii) the expiration of the waiting period (and any extension thereof) under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, and the receipt of other required approvals and clearances under applicable antitrust laws and certain foreign investment laws, as specified in the Purchase Agreement.
To the extent the Minimum Tender Condition is met and was not previously decreased, and provided that at such time Buyer directly or indirectly has acquired or controls at least one Common Share more than 90% of the then-outstanding Common Shares (excluding Common Shares held by Olink or any of its subsidiaries), Buyer will commence a process pursuant to Swedish law for the compulsory redemption of any outstanding Offer Securities held by shareholders who did not tender their securities in the Offer to obtain 100% ownership of the Company by Buyer (the “Compulsory Redemption”) in accordance with applicable laws, including the laws of Sweden.
/95
ITEM 9. THE OFFER AND LISTING
A.Offer and Listing Details
Our American Depositary Shares ("ADSs") have been listed on The Nasdaq Global Market under the trading ticker symbol “OLK” since March 25, 2021.
B.Plan of Distribution
Not applicable.
C.Markets
As noted above, the ADSs have been listed on The Nasdaq Global Market under the trading ticker symbol “OLK” since March 25, 2021. Prior to our initial public offering of ADSs in March 2021, there was no public market for our ADSs and common shares.
D.Selling Shareholders
Not applicable.
E.Dilution
Not applicable.
F.Expenses of the Issue
Not applicable.
ITEM 10. ADDITIONAL INFORMATION
A.Share Capital
Not applicable.
B.Memorandum and Articles of Association
General
We were founded as a private limited company under the laws of Sweden on December 13, 2018 under the name Goldcup 18086 AB and registered with the Swedish Companies Registration Office on January 4, 2019. Our current company name Olink Holding AB (publ) was registered with the Swedish Companies Registration Office on January 27, 2021.
We have twelve wholly owned subsidiaries, located in Sweden, the United States, the United Kingdom, the Netherlands, Germany, Japan, China, France and Singapore. The Swedish subsidiaries are Olink Finance AB, Olink OldCo AB, Olink Proteomics AB and Agrisera AB, the U.S. subsidiary is Olink Proteomics, Inc., the U.K. subsidiary is Olink Proteomics Ltd., the Dutch subsidiary is Olink Proteomics B.V., the German subsidiary is Olink Proteomics GmbH, the Japanese subsidiary is Olink Proteomics KK, the Chinese subsidiary is Olink Biotech (Shanghai) Co., Ltd., the French subsidiary is Olink Proteomics SAS and the Singaporean subsidiary is Olink Proteomics SG Pte. Ltd.
Our registered office is located at c/o Olink Proteomics AB, Salagatan 16F, SE-753 30, Uppsala, Sweden, and our telephone number is +46 (0)18 444 39 70. Our website address is www.olink.com. We have included our website address in this Annual Report solely as an inactive textual reference. The information contained on or accessible through our website is not incorporated by reference into this Annual Report.
Object of the Company
Our object is set forth in Section 3 of our Articles of Association and is to directly and indirectly develop, manufacture, market and sell biotech products and services, and to conduct other related business.
Powers of the Directors
/96
Our Board of Directors has the responsibility for our organization and the oversight of the management of our affairs. Furthermore, our Board of Directors supervises the performance of our chief executive officer and his or her actions. Our Board of Directors may exercise all powers that are not required under the Swedish Companies Act or under our Articles of Association to be exercised or taken by our shareholders.
Number of Directors
Our Articles of Association provide that our Board of Directors shall consist of three to nine members and no more than three deputy board members. Our Board of Directors currently has nine members and one deputy board member.
Rights Attached to Shares
All of the common shares have equal rights to our assets and earnings, and are entitled to one vote at the shareholders’ meeting. At the shareholders’ meeting, every shareholder may vote to the full extent of their shares held or represented, without limitation. Each common share entitles the shareholder to the same preferential rights related to issues of shares, warrants and convertible debentures relative to the number of shares they own and will have equal rights to dividends and any surplus capital upon liquidation.
Shareholders’ rights will only be changed in accordance with the procedures set out in the Swedish Companies Act. Transfers of shares will not be subject to any restrictions.
Preemptive Rights
Under the Swedish Companies Act, shareholders of any class of shares will generally have a preemptive right to subscribe for shares and other equity related securities issued of any class in proportion to their shareholdings. Shareholders will have preferential rights to subscribe for new shares in proportion to the number of shares they own. If an offering is not fully subscribed for based on subscription rights, shares may be allocated to subscribers without subscription rights. The preemptive right to subscribe does not apply in respect of shares issued paid for with non-cash consideration or of shares issued pursuant to convertible debentures or warrants previously issued by the Company.
The preemptive right to subscribe for new shares may be set aside. A share issue with deviation from the shareholders’ preemptive rights may be resolved either by the shareholders at a shareholders’ meeting, or by the Board of Directors if the board resolution is preceded by an authorization therefor from the shareholders’ meeting. A resolution to issue shares with deviation from the shareholders’ preemptive rights and a resolution to authorize the Board of Directors to do the same must be passed by two-thirds or nine-tenths (if the persons eligible to subscribe for shares are employees, directors or the CEO of the Company) of both the votes cast and the shares represented at the shareholders’ meeting resolving on the share issue or the authorization of the Board of Directors.
Voting at Shareholder Meetings
Under the Swedish Companies Act, shareholders entered into the shareholders’ register as of the record date are entitled to vote at a shareholder meeting (in person or by appointing a proxyholder). In accordance with our Articles of Association, shareholders must give notice of their intention to attend the shareholders’ meeting in accordance with the instructions of, and no later than the date specified in, the notice.
Shareholders who have their shares registered through a nominee and wish to exercise their voting rights at a shareholders’ meeting must request to be temporarily registered as a shareholder and entered into the shareholders’ register at the record date of the general meeting. The rights described herein do not apply to holders of ADSs. See Exhibit 2.5 filed to this Form 20-F for further discussion of ADSs.
Shareholder Meetings
The meeting of shareholders is our highest decision-making body and serves as an opportunity for our shareholders to make decisions regarding our affairs. Shareholders who are registered in the share register maintained by Euroclear Sweden AB six banking days before the meeting (excluding Saturdays, Sundays, Midsummer Eve, Christmas Eve, New Year’s Eve and holidays in accordance with the Swedish Public Holiday law (Sw. Lag (1989:253) om allmänna helgdagar), and nominees, who may continue to register voting rights up and until the fourth banking day before the meeting, and which shareholders and nominees have notified us no later than the date specified in the notice described below, have the right to participate at our shareholders’ meetings, either in person or by a proxyholder. All shareholders will have the same participation and voting rights at shareholders’ meetings. At the annual shareholders’ meeting, inter alia, members of the Board of Directors are elected, and a vote is held on whether each individual Board Member and the chief executive officer will be discharged from any potential liabilities for the previous fiscal year. Auditors are elected as well. Decisions
/97
are made concerning adoption of annual reports, allocation of earnings, fees for the Board of Directors and the auditors, and other essential matters that require a decision by the meeting. Most decisions require a simple majority but the Swedish Companies Act dictates other thresholds in certain instances. Shareholders will have the right to ask questions to our Board of Directors and management at shareholders’ meetings which pertain to the business of the Company and also have an item brought forward at the meeting. In order for us to include the item in the notice of the annual shareholders’ meeting, a request for an item discussion must be received by us normally seven weeks before the meeting. Any request for the discussion of an item at the annual shareholders’ meeting shall be made to the Board of Directors. The Board shall convene an extraordinary shareholders’ meeting, if shareholders who together represent at least 10% of all shares in the Company so demand in writing to discuss or resolve on a specific item or if our auditor so demands.
Shareholder meetings must be held physically. However, it is possible to allow shareholders to participate electronically at a physical shareholders' meeting. Please note that from January 1 2024, the Swedish Companies Act will allow companies to convene shareholders' meeting only through electronic means (i.e. no physical meeting is therefore required). If the Company would wish to allow electronic shareholder meetings, a provision in the Articles of Association needs to be implemented.
Notices
The Swedish Companies Act requirements for notice are described below in “- Differences in Corporate Law - Notices.”
Subject to our Articles of Association, we must publish the full notice of a shareholders’ meeting by way of press release, on our website and in the Swedish Official Gazette, and must also publish an advertisement in Svenska Dagbladet, a daily Swedish newspaper, that such notice has been published. The notice of the annual shareholders’ meeting will be published no sooner than six weeks and no later than four weeks before the date of the meeting. The notice for any extraordinary shareholders’ meeting during which a proposal to amend the Articles of Association will be addressed, must be published no sooner than six weeks and no later than four weeks before the date of the meeting. The notice must include an agenda listing each item that shall be voted upon at the meeting and a summary of each proposal that is not of minor significance for us. The notice of any other extraordinary shareholders’ meetings will be published no sooner than six weeks and no later than two weeks before the date of the meeting.
Record Date
Under the Swedish Companies Act, in order for a shareholder to participate in a shareholders’ meeting, the shareholder must have its shares registered in its own name in the share register on the sixth banking day, with the possibility for nominee registered shareholders to register voting rights up and until the fourth banking day, as described above prior to the date of the shareholders’ meeting. In accordance with section 6 of our Articles of Association, shareholders must give notice of their intention to attend the shareholders’ meeting no later than the date specified in the notice.
Amendments to the Articles of Associations
Under the Swedish Companies Act, an amendment of our Articles of Association requires a resolution passed at a shareholders’ meeting. The number of votes required for a valid resolution depends on the type of amendment; however, any amendment must be approved by not less than two-thirds of the votes cast and represented at the meeting. The Board of Directors is not allowed to make amendments to the Articles of Association absent shareholder approval.
Federal Forum Provision in the Articles of Association
Our Articles of Association provide that, unless we consent in writing to the selection of an alternative forum and without any infringement on Swedish forum provisions and without applying Chapter 7, Section 54 of the Swedish Companies Act (2005:551), the United States District Court for the Southern District of New York shall be the sole and exclusive forum for resolving any complaint filed in the United States asserting a cause of action arising under the Securities Act (Federal Forum Provision). In addition, our Articles of Association provide that any person or entity purchasing or otherwise acquiring any interest in our shares of capital stock will be deemed to have notice of and consented to the Federal Forum Provision; provided, however, that our shareholders cannot and will not be deemed to have waived our compliance with the U.S. federal securities laws and the rules and regulations thereunder.
We recognize that the Federal Forum Provision may impose additional litigation costs on shareholders in pursuing any such claims, particularly if the shareholders do not reside in or near the State of New York. Additionally, the Federal Forum Provision may limit our shareholders’ ability to bring a claim in a United States judicial forum that they find favorable for disputes with us or our directors, officers or employees, which may discourage the filing of lawsuits against us and our directors, officers and employees, even though an action, if successful, might benefit our shareholders. In addition, while the Delaware Supreme Court ruled in March 2020 that federal forum selection provisions purporting to require claims
/98
under the Securities Act be brought in federal court are “facially valid” under Delaware law, there is uncertainty as to whether other United States or Swedish courts will enforce our Federal Forum Provision. If the Federal Forum Provision is found to be unenforceable, we may incur additional costs associated with resolving such matters. The Federal Forum Provision may also impose additional litigation costs on shareholders who assert that the provision is not enforceable or invalid. The United States District Court for the Southern District of New York may also reach different judgments or results than would other courts, including courts where a shareholder considering a United States based action may be located or would otherwise choose to bring the action, and such judgments may be more or less favorable to us than our shareholders.
Provisions Restricting Change of Control of Our Company
Neither our Articles of Association nor the Swedish Companies Act contains any restrictions on change of control.
Differences in Corporate Law
The applicable provisions of the Swedish Companies Act differ from laws applicable to U.S. corporations and their shareholders. Set forth below is a summary of certain differences between the provisions of, inter alia, the Swedish Companies Act applicable to us and the Delaware General Corporation Law relating to shareholders’ rights and protections. We are not subject to Delaware law but are presenting this description for comparative purposes. This summary is not intended to be a complete discussion of the respective rights and it is qualified in its entirety by reference to Delaware law and Swedish law.
Number of Directors
Sweden. Under the Swedish Companies Act, a public limited company shall have a board of directors consisting of at least three directors of which one should be chairman. Not less than one-half of the directors shall be resident within the European Economic Area (unless otherwise approved by the Swedish Companies Registration Office). The actual number of Board members shall be determined by a shareholders’ meeting, within the limits set out in the company’s articles of association. In addition, under certain circumstances employee representatives are entitled to be represented on the Board of Directors.
Delaware. Under the Delaware General Corporation Law, a corporation must have at least one director and the number of directors shall be fixed by or in the manner provided in the bylaws. The Delaware General Corporation Law does not address director independence, though Delaware courts have provided general guidance as to determining independence, including that the determination must be both an objective and a subjective assessment.
Removal of Directors
Sweden. Under the Swedish Companies Act, directors appointed at a shareholders’ meeting may be removed by a resolution adopted at a shareholders’ meeting, upon the affirmative vote of a simple majority of the votes cast.
Delaware. Under the Delaware General Corporation Law, unless otherwise provided in the certificate of incorporation, directors may be removed from office, with or without cause, by a majority stockholder vote, though in the case of a corporation whose board is classified, stockholders may effect such removal only for cause.
Vacancies on the Board of Directors
Sweden. Under the Swedish Companies Act, if a director’s tenure should terminate prematurely, the election of a new director may be deferred until the time of the next annual shareholders’ meeting, providing there are enough remaining directors to constitute a quorum.
Delaware. Under the Delaware General Corporation Law, vacancies on a corporation’s board of directors, including those caused by an increase in the number of directors, may be filled by a majority of the remaining directors.
Annual Shareholders’ Meeting
Sweden. Under the Swedish Companies Act, within six months of the end of each fiscal year, the shareholders shall hold an annual shareholders’ meeting at which the board of directors shall present the annual report and auditor’s report and, for a parent company which is obliged to prepare group accounts, the group accounts and the auditor’s report for the group. Shareholder meetings shall be held in the city stated in the articles of association. The minutes of a shareholders’ meeting must be made available to the shareholders at the office of the company no later than two weeks after the meeting and a copy of the minutes shall be sent to those shareholders who so request and who state their postal address.
/99
Delaware. Under the Delaware General Corporation Law, the annual meeting of stockholders shall be held at such place, on such date and at such time as may be designated from time to time by the board of directors or as provided in the certificate of incorporation or by the bylaws. If a company fails to hold an annual meeting or fails to take action by written consent to elect directors in lieu of an annual meeting for a period of 30 days after the date designated for the annual meeting, or if no date was designated, 13 months after either the last annual meeting or the last action by written consent to elect directors in lieu of an annual meeting, whichever is later, the Delaware Court of Chancery may summarily order a meeting to be held upon the application of any stockholder or director. The Delaware General Corporation Law does not require minutes of stockholders’ meetings to be made public.
Special Meeting
Sweden. Under the Swedish Companies Act, the board of directors shall convene an extraordinary shareholders’ meeting if a shareholder minority representing at least ten percent of the company’s shares or the auditor of the company so demands, and the board of directors may convene an extraordinary shareholders’ meeting whenever it believes reason exists to hold an extraordinary shareholders’ meeting prior to the next annual shareholders’ meeting.
Delaware. Under the Delaware General Corporation Law, special meetings of the stockholders may be called by the board of directors or by such person or persons as may be authorized by the certificate of incorporation or by the bylaws.
Notices
Sweden. Under the Swedish Companies Act, a shareholders’ meeting must be preceded by a notice. The notice of the annual shareholders’ meeting of shareholders and a notice including a proposal to amend the articles of association of any meeting of shareholders must be issued no sooner than six weeks and no later than four weeks before the date of the meeting. In general, notice of other extraordinary shareholders’ meetings must be issued no sooner than six weeks and no later than two weeks before the date of the meeting. Public companies must always notify shareholders of a shareholders’ meeting by an announcement in the Swedish Official Gazette, and by advertisement in at least one Swedish nationwide newspaper specified in the articles of association, and by making the notice available on the company’s website.
Delaware. Under the Delaware General Corporation Law, unless otherwise provided in the certificate of incorporation or bylaws, written notice of any meeting of the stockholders must be given to each stockholder entitled to vote at the meeting not less than ten nor more than 60 days before the date of the meeting and shall specify the place, date, hour, and purpose or purposes of the meeting.
Preemptive Rights
Sweden. Under the Swedish Companies Act, shareholders of any class of shares have a preemptive right to subscribe for shares issued of any class in proportion to their shareholdings. The preemptive right to subscribe does not apply in respect of shares issued for non-cash consideration or of shares issued pursuant to convertible debentures or warrants previously issued by the company. The preemptive right to subscribe for new shares may also be set aside by a resolution passed by two thirds or nine-tenths (if the persons eligible to subscribe for shares are employees, directors or the CEO of the Company) of the votes cast and shares represented at the shareholders’ meeting resolving upon the issue.
Delaware. Under the Delaware General Corporation Law, unless otherwise provided in a corporation’s certificate of incorporation, a stockholder does not, by operation of law, possess preemptive rights to subscribe to additional issuances of the corporation’s stock.
Shareholder Vote on Certain Transactions
Sweden. In matters which do not relate to elections and are not otherwise governed by the Swedish Companies Act or the articles of association, resolutions shall be adopted at the shareholders’ meeting by a simple majority of the votes cast. In the event of a tied vote, the chairman of the shareholders meeting shall have the casting vote. For matters concerning securities of the company, such as new share issuances, and other transactions such as mergers, and a change from a public to a private company (or vice-versa), the articles of association may only prescribe thresholds which are higher than those provided in the Swedish Companies Act.
Unless otherwise prescribed in the articles of association, the person who receives the most votes in an election shall be deemed elected. In general, a resolution involving the alteration of the articles of association shall be valid only when supported by shareholders holding not less than two-thirds of both the votes cast and the shares represented at the shareholders’ meeting. The Swedish Companies Act lays out numerous exceptions for which a higher threshold applies, including restrictions on certain rights of shareholders, limits on the number of shares shareholders may vote at the
/100
shareholders’ meeting, directed share issues to directors, employees and other closely related parties, and changes in the legal relationship between shares.
Delaware. Generally, under Delaware law, unless the certificate of incorporation provides for the vote of a larger portion of the stock, completion of a merger, consolidation, sale, lease or exchange of all or substantially all of a corporation’s assets or dissolution requires: (i) the approval of the board of directors; and (ii) approval by the vote of the holders of a majority of the outstanding stock or, if the certificate of incorporation provides for more or less than one vote per share, a majority of the votes of the outstanding stock of a corporation entitled to vote on the matter.
Registration Rights Agreement
This summary may not contain all of the information about the registration rights agreement that is important to you. We urge you to read carefully the registration rights agreement in its entirety as it is the legal document governing the registration rights.
In connection with the closing of our initial public offering, we entered into a registration rights agreement with certain of our existing shareholders (for purposes of this section, the Existing Shareholders). Under this agreement, the following persons are entitled to registration rights: Knilo InvestCo AB or any of its assignees or successors (collectively, Knilo InvestCo) and the Existing Shareholders (together with Knilo InvestCo, for purposes of this section, the Holders). The summary of the material provisions of the registration rights agreement below and elsewhere in this Annual Report is qualified in its entirety by reference to the registration rights agreement, a copy of which is filed as Exhibit 2.3 to this Annual Report on Form 20-F.
Demand registration rights. At any time following the later of 180 days after our initial public offering and the expiration of the lock-up period following our initial public offering or earlier if the underwriters waive certain lock-up restrictions, we will be required to file registration statements in respect of registrable securities held by Knilo InvestCo if Knilo InvestCo so requests as follows:
•Long-Form registration. We will be required to effect an unlimited number of registrations for Knilo InvestCo on Form F-1 or Form S-1 at the request of Knilo InvestCo for all or any portion of its registrable securities (Long-Form Registration).
•Short-Form registration. After we become eligible under applicable securities laws to file a registration statement on Form F-3 or Form S-3, as applicable, which will not be until at least 12 months after the date of this Annual Report, we will be required to effect an unlimited number of registrations at the request of Knilo InvestCo on Form F-3 or Form S-3 of all or any portion of its registrable securities (Short-Form Registration, and together with a Long- Form Registration, a Demand Registration).
With respect to the above registrations, we will be required to, within three business days, give notice of a demand from Knilo InvestCo to the other Holders that will be entitled to registration rights and include their shares in the registration if they so request. If no request for inclusion from a Holder is received within three business days after we deliver a notice of such Demand Registration, such Holder shall have no further right to participate in such Demand Registration. A Holder who is, or who is controlled by any person who is, an employee of us or our subsidiaries may participate in a Demand Registration within the 12-month period immediately following the completion of our initial public offering, only if and to the extent the aggregate of (i) the registrable securities such Holder will include in such Demand Registration and (ii) the common shares such Holder has sold, transferred, assigned, distributed or otherwise conveyed prior to such Demand Registration does not exceed the 20% of the total common shares held by such Holder immediately prior to the completion of this offering (including any common shares such Holder sold in this offering, if any) (and where Knilo InvestCo will have the full and absolute discretion to determine the extent by which any cutbacks are required and which Holders will be affected), unless otherwise agreed by Knilo InvestCo.
In the event that the managing underwriter advises in good faith that the number of securities requested to be included in a Demand Registration for an underwritten offering exceeds the number that can be sold in the market in an orderly fashion, in the case of a Demand Registration, the shares to be included shall be allocated as follows: (i) in the event that Knilo InvestCo, directly or indirectly, holds more than 20% of the common shares then outstanding, first, pro rata among participating Holders in the underwritten offering, including Knilo InvestCo, on the basis of the percentage of the registrable securities owned by such Holders, and second, the securities sought to be registered by us for our own account; or (ii) in the event Knilo InvestCo, directly or indirectly, holds 20% or less of the common shares then outstanding, first, any registrable securities for which inclusion in such Demand Registration was requested by Knilo InvestCo, second, pro-rata among the participating Holders (other than Knilo InvestCo) on the basis of the percentage of the registrable securities owned by such Holders, and third, the securities sought to be registered by us for own account.
Frequency of Registrations. We will not be required to effect any Demand Registration requested during the 90-day period following the date of an underwritten offering initiated by us (other than pursuant to a registration statement on Form F-4,
/101
S-4 or S-8 or a Piggy-Back Underwritten Offering). There is no limit to the number of such registrations that Knilo InvestCo may request. We will be required to keep a Demand Registration effective for the lesser of 180 days and the time required to complete the distribution of all securities in the manner contemplated in connection with the Demand Registration. In addition, we will be able to delay effecting a Demand Registration or suspend the use of a registration statement or cease to permit the use of the Annual Report included in a Demand Registration’s registration statement in certain instances with approval of our board of directors for a “valid business reason” (as defined in the registration rights agreement) twice in any 12-month period on each occasion for a period not to exceed 90 days and for periods not to exceed 120 days in the aggregate during any 12-month period.
Piggy-back registration rights. The Holders also have the right to request the inclusion of their registrable securities in any registration statements filed by us in the future for the purposes of a public offering, subject to specified exceptions (each such offering, a Piggy-Back Underwritten Offering). A Holder may participate in a Piggy-Back Underwritten Offering only if Knilo InvestCo will participate in the same offering. In the event that the Knilo InvestCo withdraws from a Piggy-Back Underwritten Offering, all the other participating Holders will be deemed to have been withdrawn from such offering. A Holder who is, or who is controlled by any person who is, an employee of us or our subsidiaries may participate in a Piggy- Back Underwritten Offering within the 12-month period immediately following the completion of this offering, only if and to the extent the aggregate of (i) the registrable securities such Holder will include in such Piggy-Back Underwritten Offering and (ii) the common shares such Holder has sold, transferred, assigned, distributed or otherwise conveyed prior to such
Piggy-Back Underwritten Offering does not exceed the 20% of the total common shares held by such Holder immediately prior to the completion of this offering (including any common shares such Holder sold in this offering, if any) (and where Knilo InvestCo will have the full and absolute discretion to determine the extent by which any cutbacks are required and which Holders will be affected), unless otherwise agreed by the Knilo InvestCo. In the event that the managing underwriter advises in good faith that the number of shares proposed to be included exceeds the number which can be sold in the market in an orderly fashion, the shares to be included in the registration statement shall be allocated as follows: (i) in the event that Knilo InvestCo, directly or indirectly, holds more than 20% of the common shares then outstanding, first, the securities we propose to issue and sell for our own account, and second, the registrable securities requested to be included in such registration, pro rata among the participating Holders of such registrable securities on the basis of the number of registrable shares owned by each participating Holders; or (ii) in the event that Knilo InvestCo, directly or indirectly, holds 20% or less of the common shares then outstanding, first, the securities we propose to issue and sell for our own account, second, any registrable securities for which inclusion in such piggy-back registration was requested by Knilo InvestCo, and third, pro-rata among the participating Holders (other than Knilo InvestCo) on the basis of the percentage of the registrable securities owned by such participating Holders.
Termination. All registration rights granted to any Holder will terminate when no registrable securities are outstanding.
Expenses. We will pay all expenses in carrying out the above registrations, including the reasonable fees and expenses of counsel for the Holders participating in a registration as a group.
Shareholders Agreement
The summary of the material provisions of the shareholder agreement below and elsewhere in this Annual Report is qualified in its entirety by reference to the shareholder agreement, a copy of which is filed as Exhibit 2.4 to this Annual Report on Form 20-F. This summary may not contain all of the information about the shareholder agreement that is important to you. We urge you to read carefully the shareholder agreement in its entirety.
In connection with the closing of our initial public offering, we entered into a shareholder agreement with certain of our existing minority shareholders (and where relevant, their ultimate owners) (for purposes of this section, the Minority Holders) and Knilo InvestCo AB (or any of its assignees or successors) (collectively, Knilo InvestCo), under which each Minority Holder agreed to certain transfer restrictions on their shares, warrants, convertible debentures and other equity, equity- related or similar instruments of any kind (including ADSs) and any other instruments that can be converted into or given a right to subscribe or purchase any of the aforementioned instruments, and in relation to the instruments issued by us, that are not listed on a stock exchange (collectively, “equity instruments” for purposes of this section) and grant Knilo InvestCo the right to acquire their equity instruments in the event that such Minority Holder ceases to be a director, officer or employee of us (or our subsidiaries) during a certain period.
Transfer restrictions. Subject to certain permitted sales (including under the registration rights agreement), the Minority Holders (and their ultimate owners, as relevant) will not sell or otherwise dispose their equity instruments for a period of up to 12 months after the completion of our initial public offering without the prior written consent of Knilo InvestCo.
Call options. Certain of the Minority Holders will be required to offer their equity instruments for sale to Knilo InvestCo for a consideration equal to the lower of the acquisition cost and the fair market value of the relevant equity instruments if the relevant Minority Holder ceases to be a director, officer or employee of us (or our subsidiaries) during a certain period of time (generally up to 12 months after the completion of our initial public offering).
/102
Drag-along and tag-along. The Minority Holders are subject to drag-along obligations and tag-along rights on a pro rata basis with Knilo InvestCo in the case of a sale of equity instruments representing more than 50% of the votes of all equity instruments.
Power of attorney. The Minority Holders will appoint each of Knilo InvestCo (and its representatives) and the Minority Holders’ representative to vote at general meetings of our shareholders.
Termination. The shareholder agreement will terminate in relation to a Minority Holder upon such Minority Holder ceasing to hold equity instruments in us. The shareholder agreement will terminate in relation to all parties upon (i) written notice of termination by Knilo InvestCo or (ii) Knilo InvestCo (or its affiliates) ceasing to hold an interest in us.
Stock Exchange Listing
Our ADSs are listed on The Nasdaq Global Market under the symbol “OLK”.
Transfer Agent and Registrar of Shares
Our share register is maintained by Euroclear Sweden AB. The share register reflects only record owners of our common shares. Holders of the ADSs will not be treated as our shareholders and their names will therefore not be entered in our share register. The depositary, the custodian or their nominees will be the holder of the common shares underlying the ADSs. Holders of the ADSs have a right to receive the common shares underlying their ADSs subject to the terms and conditions of the deposit agreement. For discussion on the ADSs and ADS holder rights, see Exhibit 2.5 filed to this Form 20-F for further discussion of ADSs.
C.Material Contracts
Except as otherwise disclosed in this Annual Report (including the exhibits hereto), we are not currently, and have not been in the last two years, party to any material contract, other than contracts entered into in the ordinary course of our business.
D.Exchange Controls
There are currently no legal restrictions in Sweden on international capital movements and foreign exchange transactions, except in limited embargo circumstances relating to certain areas, entities or persons as a result of applicable resolutions adopted by the United Nations and the European Union. Restrictions currently exist with respect to, among others, Belarus, Russia, Crimea/Sevastapol or the non-governmental controlled areas of Ukraine in the oblasts of Donetsk, Kherson, Luhansk and Zaporizhzhia, the Democratic Republic of Congo, Guinea, Guinea-Bissau, Iran, Iraq, Lebanon, Libya, North Korea, Somalia, South Sudan, Sudan, Syria, Tunisia and Zimbabwe.
E.Taxation
The following summary contains a description of material Swedish and U.S. federal income tax consequences of the acquisition, ownership and disposition of our common shares or ADSs. This summary should not be considered a comprehensive description of all the tax considerations that may be relevant to the decision to acquire common shares or ADSs.
Material U.S. Federal Income Tax Considerations for U.S. Holders
The following is a description of certain material U.S. federal income tax considerations for U.S. Holders (defined below) with respect to their ownership and disposition of our common shares or ADSs. It is not a comprehensive description of all tax considerations that may be relevant to a particular person’s decision to acquire common shares or ADSs. This discussion applies only to a U.S. Holder that holds our common shares or ADSs as a capital asset for tax purposes (generally, property held for investment). In addition, it does not describe all of the tax consequences that may be relevant in light of a U.S. Holder’s particular circumstances, including state and local tax consequences, estate and gift tax consequences, alternative minimum tax consequences, special tax accounting rules under Section 451(b) of the Internal Revenue Code of 1986, as amended (the “Code”), the potential application of the Medicare contribution tax on net investment income, the base erosion and anti-abuse tax under Section 59A of the Code, and tax consequences applicable to U.S. Holders subject to special rules, such as:
•banks, insurance companies, and certain other financial institutions;
•certain former citizens or long-term residents of the United States;
/103
•dealers or traders in securities who use a mark-to-market method of tax accounting;
•persons holding common shares or ADSs as part of a hedging transaction, “straddle,” wash sale, conversion transaction or integrated transaction or persons entering into a constructive sale with respect to common shares or ADSs;
•persons whose “functional currency” for U.S. federal income tax purposes is not the U.S. dollar;
•brokers, dealers or traders in securities, commodities or currencies;
•tax-exempt entities or government organizations;
•a tax qualified retirement plan or other tax deferred account;
•persons holding common Shares or ADSs through entities or arrangements classified as partnerships or other pass- through entities for U.S. federal income tax purposes;
•regulated investment companies or real estate investment trusts;
•persons who acquired our common shares or ADSs pursuant to the exercise of any employee stock option or otherwise as compensation;
•persons that are resident or ordinarily resident in a jurisdiction outside the United States;
•persons holding our common shares or ADSs in connection with a trade or business, permanent establishment, or fixed base outside the United States; and
•persons who own (directly, constructively or through attribution) 10% or more (by vote or value) of our outstanding common shares or ADSs.
If an entity that is classified as a partnership for U.S. federal income tax purposes holds common shares or ADSs, the U.S. federal income tax treatment of a partner will generally depend on the status of the partner and the activities of the partnership. Partnerships holding common shares or ADSs and partners in such partnerships are encouraged to consult their tax advisors as to the particular U.S. federal income tax consequences of holding and disposing of common shares or ADSs.
The discussion is based on the Code, administrative pronouncements, judicial decisions, final, temporary and proposed Treasury Regulations, and the Convention Between the Government of the United States and the Government of Sweden for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income, signed on September 1, 1994 (the "U.S.-Sweden Tax Treaty"), all as of the date hereof, changes to any of which may affect the tax consequences described herein - possibly with retroactive effect
A “U.S. Holder” is a holder who, for U.S. federal income tax purposes, is a beneficial owner of common shares or ADSs and is:
(i)an individual who is a citizen or resident of the United States;
(ii)a U.S. domestic corporation;
(iii)an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or
(iv)a trust that (1) is subject to the primary supervision of a court within the United States and with respect to which one or more U.S. persons control all substantial decisions or (2) has a valid election to be treated as a U.S. person under applicable U.S. Treasury Regulations.
The discussion below assumes that the representations contained in the deposit agreement are true and that the obligations in the deposit agreement and any related agreement will be complied with in accordance with their terms. Generally, a holder of an ADS should be treated for U.S. federal income tax purposes as holding the common shares represented by the ADS. Consistent therewith, no gain or loss would be recognized upon an exchange of ADSs for common shares. The U.S. Treasury has expressed concerns that intermediaries in the chain of ownership between the holder of an ADS and the issuer of the security underlying the ADS could take actions that are inconsistent with the beneficial ownership of the underlying security. Therefore, actions taken by such intermediaries could affect the tax
/104
treatment of holding an ADS, including with respect to the creditability of foreign taxes, if any, and claiming a reduced tax rate, described below, on any dividends received by certain non-corporate holders.
PERSONS CONSIDERING AN INVESTMENT IN COMMON SHARES OR ADSs SHOULD CONSULT THEIR OWN TAX ADVISORS AS TO THE PARTICULAR TAX CONSEQUENCES APPLICABLE TO THEM RELATING TO THE ACQUISITION, OWNERSHIP AND DISPOSITION OF THE COMMON SHARES OR ADSs, INCLUDING THE APPLICABILITY OF U.S. FEDERAL, STATE, LOCAL AND NON-U.S. TAX LAWS.
Taxation of Distributions
Subject to the discussion below under “PFIC rules,” the gross amount of distributions (including the amount of any non-U.S. taxes withheld therefrom) paid on common shares or ADSs, other than certain pro rata distributions of common shares or ADSs, will generally be included in a U.S. Holder's income as dividend income to the extent such distribution is paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). Distributions in excess of the Company's current and accumulated earnings and profits will be treated as a non-taxable return of capital to the extent of the U.S. Holder's basis in the Shares and thereafter as capital gain. Because we do not calculate our earnings and profits under U.S. federal income tax principles, U.S. Holders should expect that distributions generally will be treated as dividends.
Dividends paid to U.S. Holders that are corporations generally will not be eligible for the dividends-received deduction generally allowed to U.S. corporations in respect of dividends received from other U.S. corporations. Subject to applicable
limitations, dividends paid to certain non-corporate U.S. Holders may be taxable at preferential rates applicable to “qualified dividend income” if we are a “qualified foreign corporation” and certain other requirements are met. However, qualified dividend income treatment will not apply if we are treated as a PFIC for our taxable year in which the dividend is paid or the preceding taxable year.
Dividends will generally be included in a U.S. Holder’s income on the date of the U.S. Holder’s receipt of the dividend. The amount of any dividend income paid in foreign currency will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder should not be required to recognize foreign currency gain or loss in respect of the dividend income. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt. Such gain or loss would generally be treated as U.S.- source ordinary income or loss. The amount of any distribution of property other than cash (and other than certain pro rata distributions of common shares or ADSs or rights to acquire common shares or ADSs) will be the fair market value of such property on the date of distribution.
Subject to generally applicable limitations and conditions, Swedish dividend withholding tax paid at the appropriate rate applicable to the U.S. Holder may be eligible for a credit against such U.S. Holder's U.S. federal income tax liability. These generally applicable limitations and conditions include new requirements recently adopted by the U.S. Internal Revenue Service ("IRS") and any Swedish tax will need to satisfy these requirements in order to be eligible to be a creditable tax for a U.S. Holder. In the case of a U.S. Holder that is eligible for, and properly elects, the benefits of the U.S.-Sweden Tax Treaty, the Swedish tax on dividends will be treated as meeting the new requirements and therefore as a creditable tax. In the case of all other U.S. Holders, the application of these requirements to the Swedish tax on dividends is uncertain and we have not determined whether these requirements have been met. If the Swedish dividend tax is not a creditable tax for a U.S. Holder or the U.S. Holder does not elect to claim a foreign tax credit for any foreign income taxes paid or accrued in the same taxable year, the U.S. Holder may be able to deduct the Swedish tax in computing such U.S. Holder's taxable income for U.S. federal income tax purposes. For foreign tax credit limitation purposes, our dividends will generally be treated as foreign source income in the passive category income basket. The rules governing foreign tax credits are complex and depend on a U.S. Holder's particular circumstances and involve the application of complex rules to those circumstances. U.S. Holders should therefore consult their tax advisors regarding the effect of the receipt of dividends for foreign tax credit limitation purposes.
/105
Sale or Other Taxable Disposition of Common Shares and ADSs
Subject to the discussion below under “PFIC rules,” gain or loss realized on the sale or other taxable disposition of common shares or ADSs will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder held the common shares or ADSs for more than one year at the time of sale or other taxable disposition. The amount of the gain or loss will equal the difference between the U.S. Holder’s tax basis in the common shares or ADSs disposed of and the amount realized on the disposition, in each case as determined in U.S. dollars. This gain or loss will generally be U.S.-source gain or loss for foreign tax credit purposes. Subject to the PFIC rules described below, the long-term capital gains recognized by certain non-corporate U.S. Holders (including individuals) will generally be subject to reduced rates of U.S. federal income tax. The deductibility of capital losses is subject to limitations.
If the consideration received by a U.S. Holder is not paid in U.S. dollars, the amount realized will be the U.S. dollar value of the payment received determined by reference to the spot rate of exchange on the date of the sale or other disposition. However, if the common shares or ADSs are treated as traded on an “established securities market” and you are either a cash basis taxpayer or an accrual basis taxpayer that has made a special election (which must be applied consistently from year to year and cannot be changed without the consent of the IRS), you will determine the U.S. dollar value of the amount realized in a non-U.S. dollar currency by translating the amount received at the spot rate of exchange on the settlement date of the sale. If you are an accrual basis taxpayer that is not eligible to or does not elect to determine the amount realized using the spot rate on the settlement date, you will recognize foreign currency gain or loss to the extent of any difference between the U.S. dollar amount realized on the date of sale or disposition and the U.S. dollar value of the currency received at the spot rate on the settlement date. Any currency gain or loss realized on the settlement date or on a subsequent conversion of the non-U.S. currency for a different U.S. dollar amount generally will be U.S. source ordinary income or loss for foreign tax credit limitation purposes. U.S. Holders should consult their tax advisors as to the U.S. federal income tax consequences of the receipt of non-U.S. currency.
PFIC Rules
A non-U.S. corporation will be classified as a passive foreign investment company, or a PFIC for any taxable year in which, after applying certain look-through rules, either:
•at least 75% of its gross income is passive income (such as interest income); or
•at least 50% of its gross assets (determined on the basis of a quarterly average) is attributable to assets that produce passive income or are held for the production of passive income.
Gross income for this purpose generally includes all sales revenue less the cost of revenue, plus income from investments and from incidental or outside operations or sources. Passive income for this purpose generally includes dividends, interest, royalties, rents and gains from commodities and securities transactions, and gains from assets that produce passive income. Cash is generally treated as an asset that produces passive income. For purposes of the PFIC income test and asset test described above, if the Company owns, directly or indirectly, 25% or more of the total value of the outstanding shares of another corporation, the Company will be treated as if it (a) held a proportionate share of the assets of such other corporation and (b) received directly a proportionate share of the income of such other corporation.
We do not believe we were classified as a PFIC during the taxable year ended December 31, 2023 and, based on the current and expected composition of our income and assets and the value of our assets, we do not expect to be a PFIC for our current taxable year. However, no assurances regarding our PFIC status can be provided for the current taxable year or any past or future taxable years. The determination of whether we are a PFIC is a fact-intensive determination made on an annual basis applying principles and methodologies that in some circumstances are unclear and subject to varying interpretation. Moreover, the value of our assets generally will be determined, in part, by reference to the market price of or common Shares and ADSs from time to time, which may fluctuate considerably. Under the income test, our status as a PFIC depends on the composition of our income which will depend on the transactions we enter into in the future and our corporate structure. U.S. Holders are urged to consult their tax advisors about the application of the PFIC rules to any of the Company's subsidiaries.
If we are classified as a PFIC in any year with respect to which a U.S. Holder owns the common shares or ADSs, we will continue to be treated as a PFIC with respect to such U.S. Holder in all succeeding years during which the U.S. Holder owns the common shares or ADSs, regardless of whether we continue to meet the tests described above unless we cease to be a PFIC and the U.S. Holder has made a “deemed sale” election under the PFIC rules. If such a deemed sale election is made, a U.S. Holder will be deemed to have sold the common shares or ADSs the U.S. Holder holds at their fair market value and any gain from such deemed sale would be subject to the rules described below. After the deemed sale election, so long as we do not become a PFIC in a subsequent taxable year, the U.S. Holder’s common shares or ADSs with respect to which such election was made will not be treated as shares in a PFIC and the U.S. Holder will not be subject to the rules described below with respect to any “excess distribution” the U.S. Holder receives from us or any gain from an actual sale or other disposition of the common shares or ADSs. U.S. Holders should consult their tax advisors as
/106
to the possibility and consequences of making a deemed sale election if we are and then cease to be a PFIC and such election is available.
For each taxable year we are treated as a PFIC with respect to U.S. Holders, U.S. Holders will be subject to special tax rules with respect to any “excess distribution” such U.S. Holder receives and any gain such U.S. Holder recognizes from a sale or other disposition (including, under certain circumstances, a pledge) of common shares or ADSs, unless (i) such U.S. Holder makes a ““qualified electing fund” election, or QEF Election, with respect to all taxable years during such U.S. Holder’s holding period in which we were a PFIC or (ii) our common shares or ADSs constitute “marketable” securities, and such U.S. Holder makes a mark- to-market election as discussed below. Distributions a U.S. Holder receives in a taxable year that are greater than 125% of the average annual distributions a U.S. Holder received during the shorter of the three preceding taxable years or the U.S. Holder’s holding period for the common shares or ADSs will be treated as an excess distribution. Under these special tax rules:
•the excess distribution or gain will be allocated ratably over a U.S. Holder’s holding period for the common shares or ADSs;
•the amount allocated to the current taxable year of disposition or distribution, and any taxable year prior to the first taxable year in which we became a PFIC, will be treated as ordinary income; and
•the amount allocated to each other year will be subject to the highest tax rate in effect for that year and the interest charge generally applicable to underpayments of tax will be imposed on the resulting tax attributable to each such year.
The tax liability for amounts allocated to years prior to the year of disposition or “excess distribution” cannot be offset by any net operating losses for such years, and gains (but not losses) realized on the sale of the common shares or ADSs cannot be treated as capital, even if a U.S. Holder holds the common shares or ADSs as capital assets. In addition, if we are a PFIC, a U.S. Holder will generally be subject to similar rules with respect to distributions we receive from, and our dispositions of the stock of, any of our direct or indirect subsidiaries that also are PFICs, as if such distributions were indirectly received by, and/or dispositions were indirectly carried out by, such U.S. Holder. U.S. Holders should consult their tax advisors regarding the application of the PFIC rules to our subsidiaries.
Certain elections exist such as a QEF Election or a mark-to-market election that may alleviate some of the adverse consequences of PFIC status and would result in an alternative treatment of a distribution on, or disposition of, our common shares or ADSs.
If a U.S. Holder makes an effective QEF Election, with respect to a PFIC, it will be taxed currently on its pro rata share of the PFIC’s ordinary earnings and net capital gain (at ordinary income and capital gain rates, respectively) for each taxable year that the entity is a PFIC, even if no distributions were received. Any distributions we make out of our earnings and profits that were previously included in such a U.S. Holder’s income under the QEF Election would not be taxable to such
U.S. Holder. Such U.S. Holder’s tax basis in its common shares would be increased by an amount equal to any income included under the QEF Election and decreased by any amount distributed on the common shares that is not included in its income. In addition, a U.S. Holder will recognize capital gain or loss on the disposition of its common shares in an amount equal to the difference between the amount realized and its adjusted tax basis in the common shares, each as determined in U.S. dollars. Once made, a QEF Election remains in effect unless invalidated or terminated by the IRS or revoked by the shareholder. A QEF Election can be revoked only with the consent of the IRS. U.S. Holders should assume that a QEF Election will not be available with respect to our common Shares or ADSs.
If a QEF Election is not in effect for the first taxable year in the U.S. Holder’s holding period in which we are a PFIC, a QEF Election generally can only be made if the U.S. Holder elects to make an applicable deemed sale or deemed dividend election on the first day of its taxable year in which the PFIC becomes a QEF pursuant to the QEF Election. The deemed gain or deemed dividend recognized with respect to such an election would be subject to the general tax treatment of PFICs discussed above.
Alternatively, U.S. Holders can avoid the interest charge on excess distributions or gain relating to the common shares or ADSs by making a mark-to-market election with respect to the common shares or ADSs, provided that the common shares or ADSs are “marketable.” Common shares or ADSs will be marketable if they are “regularly traded” on certain U.S. stock exchanges or on a foreign stock exchange that meets certain conditions. For these purposes, the common shares or ADSs will be considered regularly traded during any calendar year during which they are traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. Any trades that have as their principal purpose meeting this requirement will be disregarded. Nasdaq is a qualified exchange for these purposes. Provided the ADSs remain listed on Nasdaq and are regularly traded, and you are a holder of ADSs, we expect that the mark-to-market election would be available to you if we are a PFIC. Each U.S. Holder should consult its tax advisor as to whether a mark-to-market election is available or advisable with respect to the common shares or ADSs.
/107
A U.S. Holder that makes a mark-to-market election must include in ordinary income for each year an amount equal to the excess, if any, of the fair market value of the common shares or ADSs at the close of the taxable year over the U.S. Holder’s adjusted tax basis in the common shares or ADSs. An electing holder may also claim an ordinary loss deduction for the excess, if any, of the U.S. Holder’s adjusted basis in the common shares or ADSs over the fair market value of the common shares or ADSs at the close of the taxable year, but this deduction is allowable only to the extent of any net mark-to-market gains for prior years. Gains from an actual sale or other disposition of the common shares or ADSs will be treated as ordinary income, and any losses incurred on a sale or other disposition of the shares will be treated as an ordinary loss to the extent of any net mark-to-market gains for prior years. Once made, the election cannot be revoked without the consent of the IRS, unless the common shares or ADSs cease to be marketable.
However, a mark-to-market election generally cannot be made for equity interests in any lower-tier PFICs that we own, unless shares of such lower-tier PFIC are themselves “marketable.” As a result, even if a U.S. Holder validly makes a mark- to-market election with respect to our common shares or ADSs, the U.S. Holder may continue to be subject to the PFIC rules (described above) with respect to its indirect interest in any of our investments that are treated as an equity interest in a PFIC for U.S. federal income tax purposes.
U.S. Holders should consult their tax advisors to determine whether the mark-to-market election would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
Unless otherwise provided by the U.S. Treasury, each U.S. shareholder of a PFIC is required to file an annual report containing such information as the U.S. Treasury may require. U.S. Holders should consult their tax advisors regarding the requirements of filing such information returns under these rules.
WE STRONGLY URGE YOU TO CONSULT YOUR TAX ADVISOR REGARDING THE IMPACT OF OUR PFIC STATUS ON YOUR INVESTMENT IN THE COMMON SHARES OR ADSs AS WELL AS THE APPLICATION OF THE PFIC RULES TO YOUR INVESTMENT IN THE COMMON SHARES OR ADSs.
Information Reporting and Backup Withholding
Payments of dividends and sales proceeds that are made within the United States or through certain U.S.- related financial intermediaries generally are subject to information reporting, and may be subject to backup withholding, unless (i) the U.S. Holder is a corporation or other exempt recipient or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding on a duly executed IRS Form W-9 or otherwise establishes an exemption.
Backup withholding is not an additional tax. The amount of any backup withholding from a payment to a U.S. Holder may be credited against the U.S. Holder’s U.S. federal income tax liability and may entitle the U.S. Holder to a refund, provided that the required information is timely furnished to the IRS.
Information with Respect to Foreign Financial Assets
Certain U.S. Holders who own "specified foreign financial assets" with an aggregate value in excess of $50,000 are generally required to report information relating to the common shares or ADSs, subject to certain exceptions (including an exception for common shares or ADSs held in accounts maintained by certain U.S. financial institutions), by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. Such U.S. Holders who fail to timely furnish the required information may be subject to a penalty. Additionally, if a U.S. Holder does not file the required information, the statute of limitations with respect to tax returns of the U.S. Holder to which the information relates may not close until three years after such information is filed and may extend to six years in the case of certain omissions. U.S. Holders should consult their tax advisors regarding their reporting obligations with respect to their ownership and disposition of the common shares or ADSs.
Material Swedish Tax Considerations
The following is a summary of certain material Swedish tax issues for holders of common shares or ADSs that are not resident in Sweden for tax purposes. The summary is based on current legislation and is intended to provide general information only. The summary does not cover, inter alia, the special rules regarding tax-free dividends that may be applicable when investors hold common shares or ADSs that are deemed to be held for business purposes (for tax purposes), foreign companies conducting business through a permanent establishment in Sweden, or foreign companies that have been Swedish companies. Each person considering an investment in common shares or ADSs is advised to consult an independent tax advisor as to the tax consequences that could arise from the acquisition, ownership and disposition of the common shares or ADSs.
/108
Taxation of Dividends
For holders not resident in Sweden for tax purposes that receive dividends on common shares or ADSs of a Swedish limited liability company, Swedish withholding tax is normally withheld. The same withholding tax applies to certain other payments made by a Swedish limited liability company, such as payments as a result of redemption of shares and repurchase of shares through an offer directed to all shareholders or all holders of a certain class. The withholding tax rate is 30%. The tax rate is, however, generally reduced under an applicable tax treaty. For example, under the U.S.-Sweden Tax Treaty the tax rate on dividends paid to U.S. holders entitled to the benefits of the U.S.-Sweden Tax Treaty should not exceed 15%. In Sweden, withholding tax deductions are normally carried out by Euroclear Sweden AB or, in respect of nominee- registered shares, by the nominee. The tax treaties Sweden has entered into generally enable the withholding tax deduction to be made in accordance with the tax rate stipulated in the treaty, provided that Euroclear Sweden AB or the nominee, as applicable, has received the required information concerning the tax residency of the investor entitled to the dividend (this applies also under the U.S.-Sweden Tax Treaty). Furthermore, investors entitled to reduced tax rates under applicable tax treaties may claim a refund from the Swedish tax authorities within five calendar years following the year the dividend was distributed if the full withholding tax rate at 30% has been withheld.
Taxation of Capital Gains
Holders not resident in Sweden for tax purposes are normally not liable for capital gains taxation in Sweden upon disposals of common shares or ADSs. Holders of common shares or ADSs may, however, be subject to taxation in their state of residence.
According to a special rule, private individuals not resident in Sweden for tax purposes are, however, subject to Swedish capital gains taxation upon disposals of common shares or ADSs if they have been residents of Sweden due to a habitual abode in Sweden or a stay in Sweden for six consecutive months at any time during the calendar year of disposal or the ten calendar years preceding the year of disposal. In a number of cases though, the applicability of this rule is limited by tax treaties. The applicability of this rule may be limited under the U.S.-Sweden Tax Treaty.
F.Dividends and Paying Agents
Not applicable.
G.Statement by Experts
Not applicable.
H.Documents on Display
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers. Accordingly, we are required to file reports and other information with the SEC, including annual reports on Form 20-F and reports on Form 6-K. The SEC maintains a website at www.sec.gov that contains reports and other information regarding registrants that make electronic filings with the SEC using its EDGAR system. Our filings made with the SEC are available on the SEC’s website. We also make available on the investor relations section of our website, free of charge, our annual reports on Form 20-F and our reports on Form 6-K, including any amendments to these reports, as well as certain other SEC filings, as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. Our website address is www.olink.com. We have included our website address in this Annual Report solely as an inactive textual reference. The information contained on or accessible through our website is not incorporated by reference into this Annual Report.
As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act.
We will send the depositary a copy of all notices of shareholders meetings and other reports, communications and information that are made generally available to shareholders. The depositary has agreed to mail to all holders of ADSs a notice containing the information (or a summary of the information) contained in any notice of a meeting of our shareholders received by the depositary and will make available to all holders of ADSs such notices and all such other reports and communications received by the depositary. All notices of shareholders meetings and other reports, communications and information that are made generally available to shareholders are also filed or furnished with the SEC on its EDGAR system and posted to the investor relations section of our website.
/109
I.Subsidiary Information
Subsidiary information is provided in Note 10 in the Financial Statements.
J.Annual Report to Security Holders
We are furnishing our Annual Report for 2023 on Form 6-K concurrent with the filing of this Form 20-F.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
The Group's activities are subject to several financial risks: market risk (including currency risk and interest rate risk), credit risk and liquidity risk. The Group strive to minimize potential unfavorable effects from these risks on the Group’s financial results.
The aim of the Group´s financial operations is to:
•Ensure that the Group can meet its financial obligations timely,
•Manage financial risks, and
•Ensure a supply of necessary financing.
The Group´s risk management is predominantly controlled by senior management. For more details refer to Note 4.1 in the Notes to the Consolidated Financial Statements contained herein.
Market risk - Currency risk (transaction risk)
The Group operates internationally and are exposed to currency risk where invoicing is made in a currency other than the functional currency of the relevant Group entity. Mitigation of this risk occurs naturally by matching expenses and obtaining borrowings, as required, in the same foreign currency. The currency risk is monitored on a regular basis. The Group has not entered into derivative currency instruments during the reported period.
See Note 4.1 in the Notes to the Consolidated Financial Statements contained herein for a sensitivity analysis on foreign exchange risk.
Market risk - Interest-rate risk
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates.
As of December 31, 2023, the Group does not have any outstanding debt or other debt structures other than leasing. The Group does not hold any fixed-income investments.
Interest rate derivative instruments were not used during the reported periods.
Credit risk
Credit risk is the risk that a counterparty will not meet its obligations under a financial instrument or customer contract, leading to a financial loss. The Group is exposed to credit risk from its operating activities (primarily trade receivables) and from its investing activities, including deposits with banks and financial institutions and foreign exchange transactions. Credit risk relates primarily to customer credit limits, which are subject to certain credit rating rules and authorization processes. However, the majority of the customer base tends to be blue chip global companies and therefore such customers usually have strong credit ratings. Our sales are concentrated such that 43% of sales in 2023 and 45% of sales in 2022 are with customers based in the U.S.
The maximum default risk for the Group is equivalent to the net receivables reported in the Consolidated Financial Statements. The Group have historically almost non-existent credit losses and based on historical data together with a forward-looking assessment, the 2023 expected credit loss for trade receivables is disclosed in Note 18, ‘Trade receivables’.
The Group´s cash at bank is held in Investment Grade credit rated banks. To mitigate the counterparty risk cash is distributed among different banks and the cash position and distribution is monitored on a regular basis.
Other financial assets at amortized cost include rental deposits. The credit risk for other financial assets at amortized cost as at December 31, 2023 and 2022 is not material and no credit loss reserve has been recognized.
/110
Liquidity risk
Cash at bank allows the Group to meet its liquidity risk obligations as they come due. Following the Initial Public Offering of the Group in March, 2021 the liquidity risk has been managed by cash at bank deposits.
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
A.Debt Securities
Not applicable.
B.Warrants and Rights
Not applicable.
C.Other Securities
Not applicable.
/111
D.American Depositary Shares
Fees and Expenses
| Persons depositing or withdrawing shares or ADS holders must pay | For | |||||||
| $5.00 (or less) per 100 ADSs (or portion of 100 ADSs) | Issuance of ADSs, including issuances resulting from a distribution of shares or rights or other property Cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates | |||||||
| Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes | Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes | |||||||
| $.05 (or less) per ADS | Any cash distribution to ADS holders | |||||||
| A fee equivalent to the fee that would be payable if securities distributed to you had been shares and the shares had been deposited for issuance of ADSs | Distribution of securities distributed to holders of deposited securities (including rights) that are distributed by the depositary to ADS holders | |||||||
| $.05 (or less) per ADS per calendar year | Depositary services | |||||||
| Registration or transfer fees | Transfer and registration of shares on our share register to or from the name of the depositary or its agent when you deposit or withdraw shares | |||||||
| Expenses of the depositary | Cable (including SWIFT) and facsimile transmissions (when expressly provided in the deposit agreement) Converting foreign currency to U.S. dollars | |||||||
| Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes | As necessary | |||||||
| Any charges incurred by the depositary or its agents for servicing the deposited securities | As necessary | |||||||
The depositary collects its fees for delivery and surrender of ADSs directly from investors depositing shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions or by directly billing investors or by charging the book-entry system accounts of participants acting for them. The depositary may collect any of its fees by deduction from any cash distribution payable (or by selling a portion of securities or other property distributable) to ADS holders that are obligated to pay those fees. The depositary may generally refuse to provide fee- attracting services until its fees for those services are paid.
From time to time, the depositary may make payments to us to reimburse us for costs and expenses generally arising out of establishment and maintenance of the ADS program, waive fees and expenses for services provided to us by the depositary or share revenue from the fees collected from ADS holders. In performing its duties under the deposit agreement, the depositary may use brokers, dealers, foreign currency dealers or other service providers that are owned by or affiliated with the depositary and that may earn or share fees, spreads or commissions.
/112
The depositary may convert currency itself or through any of its affiliates, or the custodian or we may convert currency and pay U.S. dollars to the depositary. Where the depositary converts currency itself or through any of its affiliates, the depositary acts as principal for its own account and not as agent, advisor, broker or fiduciary on behalf of any other person and earns revenue, including, without limitation, transaction spreads, that it will retain for its own account. The revenue is based on, among other things, the difference between the exchange rate assigned to the currency conversion made under the deposit agreement and the rate that the depositary or its affiliate receives when buying or selling foreign currency for its own account. The depositary makes no representation that the exchange rate used or obtained by it or its affiliate in any currency conversion under the deposit agreement will be the most favorable rate that could be obtained at the time or that the method by which that rate will be determined will be the most favorable to ADS holders, subject to the depositary’s obligation to act without negligence or bad faith. The methodology used to determine exchange rates used in currency conversions made by the depositary is available upon request. Where the custodian converts currency, the custodian has no obligation to obtain the most favorable rate that could be obtained at the time or to ensure that the method by which that rate will be determined will be the most favorable to ADS holders, and the depositary makes no representation that the rate is the most favorable rate and will not be liable for any direct or indirect losses associated with the rate. In certain instances, the depositary may receive dividends or other distributions from us in U.S. dollars that represent the proceeds of a conversion of foreign currency or translation from foreign currency at a rate that was obtained or determined by us and, in such cases, the depositary will not engage in, or be responsible for, any foreign currency transactions and neither it nor we make any representation that the rate obtained or determined by us is the most favorable rate and neither it nor we will be liable for any direct or indirect losses associated with the rate.
Payment of Taxes
Stockholders will be responsible for any taxes or other governmental charges payable on your ADSs or on the deposited securities represented by any of your ADSs. The depositary may refuse to register any transfer of your ADSs or allow you to withdraw the deposited securities represented by your ADSs until those taxes or other charges are paid. It may apply payments owed to you or sell deposited securities represented by your ADSs to pay any taxes owed and you will remain liable for any deficiency. If the depositary sells deposited securities, it will, if appropriate, reduce the number of ADSs to reflect the sale and pay to ADS holders any proceeds, or send to ADS holders any property, remaining after it has paid the taxes.
PART II
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
Not applicable.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
Not applicable.
/113
ITEM 15. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has performed an evaluation of the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act) as of December 31, 2023, as required by Rule 13a-15(b) under the Exchange Act. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of December 31, 2023, our disclosure controls and procedures were not effective as of such date due to the material weaknesses in internal control over financial reporting, described below.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) of the Exchange Act). Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2023. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control—Integrated Framework (2013). Based on our assessment, our management concluded that our internal control over financial reporting was not effective as of December 31, 2023, due to the material weaknesses in internal control over financial reporting described below.
Material weaknesses
Based on this evaluation, our management have identified material weaknesses for the year ended December 31, 2023 and remediated a material weakness that was previously identified for the years ended December 31, 2020, 2021 and 2022.
As defined in standards established by the PCAOB, a material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of a company’s annual or interim financial statements will not be prevented or detected on a timely basis.
A material weakness with respect to internal controls was identified in our U.S. subsidiary. Local management does not timely and consistently execute and document procedures performed at material processes due to insufficient personnel with the appropriate level of internal controls skillset.
This material weakness identified with respect to the U.S. subsidiary only pertains to 2023 due to the overall business size being smaller and less complex in 2022, as sales activities and inventory were managed from Sweden for the majority of 2022.
In the year ended December 31, 2023, we identified a material weakness in our internal control environment with respect to the inventory process in Sweden. We did not design and maintain effective controls over the completeness, accuracy and existence of inventory. This is due to inconsistent application of policies and procedures during and in preparation of the physical stock count. Despite our best efforts and significant progress in this area during 2023, our testing determined that this material weakness was not fully remediated at December 31, 2023.
As of and for the years ended December 31, 2020, December 31, 2021, and December 31, 2022, we identified a material weakness relating to our technology access and change control environment not supporting an efficient or effective internal controls framework. Remediation efforts relating to this material weakness were intensified during 2023 and included (i) continuous formalization of access and change controls to our systems and identification of compensating measures, and (ii) improving governance procedures, including providing internal training in relation to our information technology policies and procedures. As a result of these efforts, our testing procedures confirmed that this material weakness was remediated at December 31, 2023.
Remediation Plan
To remedy the material weakness at our U.S. subsidiary, we have initiated several measures to improve the affected processes. Key activities include (i) formalization of execution of controls and procedures, (ii) hiring additional resources with appropriate expertise to effectively operate and document the process related internal controls and (iii) providing specific training to existing personnel.
/114
To remedy the material weakness over inventory, we have initiated several measures to improve the affected inventory process. Key activities include (i) implementing enhanced review and control procedures and policies ensuring complete and accurate physical stock count and related internal controls, and (ii) providing training to existing personnel.
The effectiveness of any system of internal control over financial reporting is subject to inherent limitations, including the exercise of judgment in designing, implementing, operating, and evaluating the controls and procedures, and the inability to eliminate misconduct completely. Accordingly, any system of internal control over financial reporting can only provide reasonable, not absolute, assurances. In addition, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. We intend to continue to monitor and upgrade our internal controls as necessary or appropriate for our business.
Attestation Report of the Registered Public Accounting Firm
Please see the report of Ernst & Young AB, an independent registered public accounting firm, included in “Item 18. Financial Statements".
Changes in internal control over financial reporting
Except for improvements to our internal control over financial reporting that are being carried out to remediate the material weaknesses described above, no change to our internal control over financial reporting occurred during the year ended December 31, 2023, that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
/115
ITEM 16. [RESERVED]
ITEM 16A. AUDIT COMMITTEE FINANCIAL EXPERT
Our Audit Committee consists of Solange Bullukian, Mary Reumuth and Robert Schueren. Solange Bullukian is the chair of the Audit Committee. Our Board of Directors has determined that all of the members of the Audit Committee satisfy the “independence” requirements set forth in Rule 10A-3 under the Exchange Act and the Nasdaq listing standards. Our Board has determined that Solange Bullukian qualifies as an “audit committee financial expert” as that term is defined under the Exchange Act.
ITEM 16B. CODE OF ETHICS
We have adopted a Code of Conduct that applies to our chief executive officer and all senior financial officers of our Company, including the chief financial officer, chief accounting officer or controller, or persons performing similar functions. The Code of Conduct is publicly available on our website at https://investors.olink.com/corporate-governance/governance-overview. We have included our website address in this Annual Report solely as an inactive textual reference. The information contained on or accessible through our website is not incorporated by reference into this Annual Report.
ITEM 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
We retained Ernst & Young AB (EY) as our independent registered public accounting firm for 2023. Set forth below is a summary of the fees paid to Ernst & Young AB for services provided in fiscal year 2023 and 2022.
| Amounts in thousands of USD | Fiscal Year 2023 | Fiscal Year 2022 | ||||||
| Audit fees | $ | 2,554 | $ | 988 | ||||
| Audit-related fees | 122 | 154 | ||||||
| All other fees | 491 | 241 | ||||||
| Tax fees | 10 | 31 | ||||||
Total remuneration Ernst & Young AB | $ | 3,177 | $ | 1,414 | ||||
All other fees in fiscal year 2023, in above summary, mainly refers to services assistance for Olink Insight which is a cloud based platform for the Olink community aimed to accelerate proteomics.
For 2021 Öhrlings PricewaterhouseCoopers AB was our independent registered public accounting firm. Set forth below is a summary of the fees paid to Öhrlings PricewaterhouseCoopers AB for services provided in fiscal year 2023 and 2022.
| Amounts in thousands of USD | Fiscal Year 2023 | Fiscal Year 2022 | ||||||
| Audit fees | $ | 22 | $ | 86 | ||||
| Audit-related fees | — | 83 | ||||||
| All other fees | 897 | 168 | ||||||
| Tax fees | 20 | 12 | ||||||
Total remuneration Öhrlings PricewaterhouseCoopers AB | $ | 939 | $ | 349 | ||||
Audit-related fees in fiscal year 2022 mainly refers to service assistance for the public offering in January 2023.
Pre-Approval Policies and Procedures
Our Audit Committee has adopted policies and procedures for the pre-approval of all auditing services and the terms thereof and non-audit services other than non-audit services prohibited under Section 10A(g) of the Exchange Act or the applicable rules of the SEC or the Public Company Accounting Oversight Board (PCAOB) to be provided to the Company by the independent auditors. However, the pre-approval requirement is waived with respect to the provision of non-audit services for the Company if the “de minimus” provisions of Section 10A(i)(1)(B) of the Exchange Act are satisfied. All non-audit services in 2022 and 2023 were pre-approved by the Audit Committee.
ITEM 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
/116
ITEM 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS
None.
ITEM 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
Not applicable.
ITEM 16G. CORPORATE GOVERNANCE
We are a “foreign private issuer” as defined by the SEC. The Sarbanes-Oxley Act of 2002, as well as related rules subsequently implemented by the SEC, requires foreign private issuers to comply with various corporate governance practices. Also as a result of being a foreign private issuer, in accordance with Nasdaq listing requirements, we may rely on home country governance requirements and certain exemptions thereunder rather than complying with all Nasdaq corporate governance standards for domestic issuers. While we voluntarily follow most Nasdaq corporate governance rules, we may choose to take advantage of the following limited exemptions:
•Exemption from filing quarterly reports on Form 10-Q containing unaudited financial and other specified information or current reports on Form 8-K upon the occurrence of specified significant events;
•Exemption from Section 16 rules requiring insiders to file public reports of their securities ownership and trading activities and providing for liability for insiders who profit from trades in a short period of time;
•Exemption from the Nasdaq requirement necessitating disclosure of any waivers of the Code of Conduct for directors and executive officers;
•Exemption from the requirement to obtain shareholder approval for certain issuances of securities, including shareholder approval of share option plans;
•Exemption from the requirement that our Audit Committee have review and oversight responsibilities over all “related party transactions,” as defined in Item 7.B of Form 20-F;
•Exemption from the requirement that our Board of Directors have a compensation committee that is composed entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities and
•Exemption from the requirement to have independent director oversight of director nominations.
/117
Furthermore, Nasdaq Rule 5615(a)(3) provides that a foreign private issuer may rely on home country corporate governance practices in lieu of certain of the rules in the Nasdaq Rule 5600 Series and Rule 5250(d). We follow Swedish corporate governance practices in lieu of Nasdaq corporate governance requirements as follows:
•We do not follow Nasdaq Rule 5620(c) regarding quorum requirements applicable to meetings of shareholders. Such quorum requirements are not required under Swedish law. The Swedish Companies Act (SFS 2005:551) and our Articles of Association provide alternative quorum requirements that are generally applicable to meetings of shareholders.
•We do not follow Nasdaq Rule 5605(b)(2), which requires that independent directors regularly meet in executive sessions where only independent directors are present. Our independent directors may choose to meet in executive sessions at their discretion.
•We do not follow Nasdaq Rule 5605(d) regarding the composition of the Remuneration Committee.
•We do not follow Nasdaq Rule 5605(e) regarding the composition of the Nominating Committee.
Although we may rely on certain home country corporate governance practices, we must comply with Nasdaq’s Notification of Noncompliance requirement (Nasdaq Rule 5625) and the Voting Rights requirement (Nasdaq Rule 5640). Further, we must have an Audit Committee that satisfies Nasdaq Rule 5605(c)(3), which addresses Audit Committee responsibilities and authority and requires that the Audit Committee consist of members who meet the independence requirements of Nasdaq Rule 5605(c)(2)(A)(ii).
As a foreign private issuer, our directors and executive officers are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the Exchange Act. They are, however, subject to the obligations to report changes in securities ownership under Section 13 of the Exchange Act and related SEC rules.
ITEM 16H. MINE SAFETY DISCLOSURE
Not applicable.
ITEM 16I. DISCLOSURES REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
ITEM 16J. INSIDER TRADING POLICIES
Not applicable.
ITEM 16K. CYBERSECURITY
The Company has implemented security measures and formal, dedicated enterprise security programs to minimize potential impact related to cyber security incidents. The Company has adopted and will continue to update policies and procedures for assessing, identifying, and managing material risks from cybersecurity threats to provide protections against such attacks in the future as well as for reporting potential cybersecurity incidents to the Chief Information Security Officer (CISO) and, if necessary, to the Audit Committee. The Company has purchased cybersecurity insurance as protection in the future. Olink engages consultants from time to time to assist with specific parts of its cybersecurity program, such as advising on cybersecurity incident response procedures, assessments exercises (e.g. table-top), penetration testing and other technical specialists, but does not otherwise engage or rely upon external assessors, consultants, auditors, or other third parties supporting the process around information systems risk mitigation.
In January 2024, the Company’s information security team determined that an unidentified threat actor was able to gain temporary access to an Olink employee’s email account via a phishing email. Based on forensic analysis, the threat actor viewed certain emails that included salary information of certain employees and certain 2023 sales results by customer account, but the Company was unable to determine if any emails or files were downloaded. Such cybersecurity incident was reported to the Company’s Audit Committee and senior management as well as the Swedish Authority for Privacy Protection. In light of the successful phishing attack, the Company determined that it is possible that the reputational harm resulting from such cybersecurity incident could have a material effect on the Company’s business strategy with customers and employees if such information is released in the future. The Company has not seen any such effects to date.
/118
Except as described above, at the time of this report, no risks from cybersecurity threats, including as a result of any previous cybersecurity incidents, have materially affected or the registrant, including its business strategy, results of operations, or financial condition. However, there are no assurances that electronic break-ins, computer viruses and similar disruptive problems, and/or sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data will not occur. For more information on cybersecurity risks, please refer to the section entitled, "ITEM 3 - KEY INFORMATION - D. Risk Factors" in this Form 20-F.
Each Company employee has a responsibility to report any identified risk or unresolved vulnerabilities to the CISO. The CISO is certified within industry standards, including Certified Information Systems Security Professional (CISSP) by the International Information System Security Certification Consortium, Certified Information Security Manager (CISM) by the Information Systems Audit and Control Association ( ISACA), Certified in Risk and Information Systems Control (CRISC) by ISACA and Certified in the Governance of Enterprise IT (CGEIT) by ISACA, and has over ten years as a security practitioner and almost twenty years of experience with IT and technology, business development and project management. In addition, the Company’s Enterprise Risk Manager has over a year of experience in enterprise risk management.
Any risks identified as high risk are escalated to the enterprise risk management process through the Chief Operating Officer, the CISO, business leaders or system owners and are assessed by senior management as part of the overall companywide risk evaluation process. Senior management plays a key role in assessing and managing cybersecurity-related risks and implementing the company’s cybersecurity policies and procedures. The Enterprise Risk Manager is responsible for aggregating, consolidating, and reporting key cybersecurity risks identified by the organization to the Group Management Team in an annual companywide risk workshop. Based on the workshop a Group Top Risk Summary Report is presented to the Audit Committee and the Board of Directors. The Group Management Team regularly monitors progress of identified and planned risk remediation activities and status of the companywide top risks through business reviews. Any material deviations is communicated to the Audit Committee.
The Audit Committee acts on behalf of the Board in fulfilling the Board’s oversight responsibility with respect to the Company’s information systems use and protection, including but not limited to data governance, privacy, compliance and cybersecurity as well as receiving and evaluating reports of potential cybersecurity incidents by the CISO, consistent with the Company’s current policies and procedures regarding the same.
PART III
ITEM 17. FINANCIAL STATEMENTS
We have elected to furnish financial statements and related information specified in Item 18.
ITEM 18. FINANCIAL STATEMENTS
See the financial statements beginning on page F-1.
ITEM 19. EXHIBITS
/119
/120
| 101.INS* | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |||||||
| 101.SCH* | Inline XBRL Taxonomy Extension Schema Document. | |||||||
/121
| 101.CAL* | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | |||||||
| 101.DEF* | Inline XBRL Taxonomy Extension Definition Linkbase Document. | |||||||
| 101.LAB* | Inline XBRL Taxonomy Extension Label Linkbase Document. | |||||||
| 101.PRE* | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | |||||||
| 104* | Cover Page Interactive Data File (formatted in Inline XBRL and contained in Exhibit 101). | |||||||
*Filed herewith.
** Furnished herewith.
# Indicates a management contract or any compensatory plan, contract or arrangement.
+ Certain portions of this exhibit have been omitted because such portions are not material and are treated by the Registrant as private or confidential and would likely cause competitive harm to the Registrant if disclosed.
/122
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| OLINK HOLDING AB (PUBL) | |||||
| By: | /s/ Jon Heimer | ||||
| Name: | Jon Heimer | ||||
| Title: | Chief Executive Officer | ||||
Date: March 25, 2024
/123
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Olink Holding AB (publ)
Opinion on the Financial Statements
We have audited the consolidated statement of income and comprehensive income, of changes in equity and of cash flows of Olink Holding AB (publ) and its subsidiaries (the “Company”) for the year ended December 31, 2021 , including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the results of operations and cash flows of the Company for the year ended December 31, 2021 in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ Öhrlings PricewaterhouseCoopers AB
March 17, 2022
We served as the Company's auditor from 2016 to 2022
F-2
Reports of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Olink Holding AB (publ)
Opinion on the Financial Statements
We have audited the accompanying consolidated statement of financial position of Olink Holding AB (publ) and subsidiaries (the “Company”) as of December 31, 2023 and 2022, the related consolidated statements of income and other comprehensive income, changes in equity and cash flows for each of the two years in the period ended December 31, 2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2023 in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 25, 2024, expressed an adverse opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
F-3
Inventory valuation of new antibodies | ||||||||
Description of the Matter | As of December 31, 2023, the Company’s inventory of $66,436 thousand, included a material amount related to new antibodies subject to research and development. As more fully described in footnote 2.3 of the Consolidated Financial Statements, inventory is stated at the lower of cost or net realizable value. When the Company develops new antibodies for their Kit products, management must estimate an expected success rate, which is utilized in their determination of the development costs capitalized as inventory Auditing the valuation of Inventory related to new antibodies is complex, due to the significant judgements and subjectivity involved in management’s estimate of the expected success rate. The significant estimation uncertainty was primarily due to the non-financial (technical feasibility) basis of the estimated success rate. Changes in the expected success rate can have a significant impact on the valuation of inventory. | |||||||
How We Addressed the Matter in Our Audit | To test the expected success rate, our audit procedures included, among others, benchmarking against historical information, where we compared the expected success rate of new antibodies against the expected success rate previously applied in early-stage testing for similar antibodies, which have completed internal testing of technical feasibility. To assess management’s ability to estimate the expected success rate, we compared the final success rate of new antibodies, which have completed internal testing of technical feasibility, to the expected success rate for the same antibodies during their early-stage testing. Our procedures also included inquiry of research and development personnel regarding the basis for the expected success rate. We tested the completeness and accuracy of underlying data used, by comparing the testing result against testing protocol | |||||||
/s/ Ernst & Young AB
We have served as the Company’s auditor since 2022.
Stockholm, Sweden
March 25, 2024
F-4
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Olink Holding AB (publ)
Opinion on Internal Control Over Financial Reporting
We have audited Olink Holding AB (publ) and subsidiaries’ internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, because of the effect of the material weaknesses described below on the achievement of the objectives of the control criteria, Olink Holding AB (publ) and subsidiaries (the Company) has not maintained effective internal control over financial reporting as of December 31, 2023, based on the COSO criteria.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. The following material weaknesses has been identified and included in management’s assessment. Management has identified material weaknesses in controls related to the U.S. subsidiary and the inventory process in Sweden.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated statement of financial position of the Company as of December 31, 2023 and 2022, the related consolidated statements of income and other comprehensive income, changes in equity and cash flows for each of the two years in the period ended December 31, 2023, and the related notes and our report dated March 25, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
F-5
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young AB
March 25, 2024
F-6
CONSOLIDATED STATEMENTS OF INCOME AND OTHER COMPREHENSIVE INCOME
| Amounts in thousands of USD | Note | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||||
| Revenue | 5 | $ | $ | $ | ||||||||||
| Cost of revenue | 6 | ( | ( | ( | ||||||||||
| Gross profit | ||||||||||||||
| Selling expenses | 6 | ( | ( | ( | ||||||||||
| Administrative expenses | 6 | ( | ( | ( | ||||||||||
| Research and development expenses | 6 | ( | ( | ( | ||||||||||
| Other operating income | ||||||||||||||
| Other operating expense | ( | ( | ( | |||||||||||
| Operating loss | ( | ( | ( | |||||||||||
| Interest income | 8 | |||||||||||||
| Interest expense | 8 | ( | ( | ( | ||||||||||
| Foreign exchange gain | 8 | |||||||||||||
| Other financial income/(expense) | 8 | ( | ||||||||||||
| Loss before tax | ( | ( | ( | |||||||||||
| Income tax benefit | 9 | |||||||||||||
| Net loss for the period (Attributable to shareholders of the Company) | $ | ( | $ | ( | $ | ( | ||||||||
| Other comprehensive (loss)/income: | ||||||||||||||
| Items that may be reclassified to profit or loss: | ||||||||||||||
| Exchange differences from translation of foreign operations | ( | ( | ||||||||||||
| Other comprehensive (loss)/income for the, period, net of tax | ( | ( | ||||||||||||
| Total comprehensive (loss)/income for the period, net of tax | $ | ( | $ | ( | $ | ( | ||||||||
| Total comprehensive (loss)/income for the period (Attributable to shareholders of the company) | $ | ( | $ | ( | $ | ( | ||||||||
| Basic and diluted loss per share | 24 | $ | ( | $ | ( | $ | ( | |||||||
The accompanying notes are an integral part of the Consolidated Financial Statements.
F-7
CONSOLIDATED STATEMENT OF FINANCIAL POSITION
| Amounts in thousands of USD | Note | As of December 31, 2023 | As of December 31, 2022 | ||||||||
| ASSETS | |||||||||||
| Non-current assets | |||||||||||
| Goodwill | 12 | $ | $ | ||||||||
| Intangible assets | 12 | ||||||||||
| Property, plant and equipment | 13 | ||||||||||
| Right-of-use assets | 14 | ||||||||||
| Deferred tax assets | 9 | ||||||||||
| Other non-current receivables | 16 | ||||||||||
| Current assets | |||||||||||
| Inventories | 17 | ||||||||||
| Trade receivables | 18 | ||||||||||
| Other receivables | 19 | ||||||||||
| Prepaid expenses and accrued income | |||||||||||
| Cash at bank and in hand | |||||||||||
| TOTAL ASSETS | $ | $ | |||||||||
| EQUITY | |||||||||||
| Share capital | 20 | ||||||||||
| Other contributed capital | 20 | ||||||||||
| Reserves | ( | ( | |||||||||
| Accumulated losses | ( | ( | |||||||||
| Total equity attributable to shareholders of the Company | $ | $ | |||||||||
| LIABILITIES | |||||||||||
| Non-current liabilities | |||||||||||
| Lease liabilities | 14, 15 | ||||||||||
| Deferred tax liabilities | 9 | ||||||||||
| Current liabilities | |||||||||||
| Lease liabilities | 14, 15 | ||||||||||
| Accounts payable | |||||||||||
| Current tax liabilities | 9 | ||||||||||
| Other current liabilities | 22 | ||||||||||
| Total liabilities | $ | $ | |||||||||
| TOTAL EQUITY AND LIABILITIES | $ | $ | |||||||||
The accompanying notes are an integral part of the Consolidated Financial Statements.
F-8
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
| Amounts in thousands of USD | Note | Share Capital | Other Contributed capital | Reserves | Accumulated loss | Total equity | ||||||||||||||
| At January 1, 2021 | $ | $ | $ | $ | ( | $ | ||||||||||||||
| Loss for the period | — | — | — | ( | ( | |||||||||||||||
| Other comprehensive income for the Period | — | — | ( | — | ( | |||||||||||||||
| Total comprehensive income/(loss) for the Period | — | — | ( | ( | ( | |||||||||||||||
| Transactions with shareholders in their role as owners | ||||||||||||||||||||
| Shareholders’ contributions | — | — | — | |||||||||||||||||
| New share issue | 20 | — | — | |||||||||||||||||
| Share based compensation program | 21 | $ | $ | |||||||||||||||||
| At December 31, 2021 | $ | $ | $ | $ | ( | $ | ||||||||||||||
| Loss for the period | — | — | — | ( | ( | |||||||||||||||
| Other comprehensive loss for the Period | — | — | ( | — | ( | |||||||||||||||
| Total comprehensive loss for the Period | — | — | ( | ( | ( | |||||||||||||||
| Transactions with shareholders in their role as owners | ||||||||||||||||||||
| Shareholders' contributions | — | — | — | — | — | |||||||||||||||
| New share issue | 20 | — | — | |||||||||||||||||
| Share based compensation program | 21 | — | — | — | ||||||||||||||||
| At December 31, 2022 | $ | $ | $ | ( | $ | ( | $ | |||||||||||||
| Loss for the period | — | — | — | ( | ( | |||||||||||||||
| Other comprehensive profit/(loss) for the Period | — | — | — | |||||||||||||||||
| Total comprehensive profit/(loss) for the Period | — | — | ( | ( | ||||||||||||||||
| Transactions with shareholders in their role as owners | ||||||||||||||||||||
| Shareholders' contributions | — | — | — | — | — | |||||||||||||||
| New share issue | 20 | — | — | |||||||||||||||||
| Share based compensation program | 21 | — | — | — | ||||||||||||||||
| At December 31, 2023 | $ | $ | $ | ( | $ | ( | $ | |||||||||||||
The accompanying notes are an integral part of the Consolidated Financial Statements.
F-9
CONSOLIDATED STATEMENT OF CASH FLOWS
| Amounts in thousands of USD | Note | For the year ended December 31, 2023 | For the year ended December 31, 2022 | For the year ended December 31, 2021 | ||||||||||
| Operating activities | ||||||||||||||
| Loss before tax | $ | ( | $ | ( | $ | ( | ||||||||
| Adjustments reconciling loss before tax to operating cash flows: | ||||||||||||||
| Depreciation and amortization | 13 | |||||||||||||
| Net finance (income)/expense | 8 | ( | ( | |||||||||||
| Loss on disposal of assets | ||||||||||||||
| Share based payment expense | 21 | |||||||||||||
| Other | ( | |||||||||||||
| Changes in working capital: | ||||||||||||||
| (Increase) in inventories | 17 | ( | ( | ( | ||||||||||
| (Increase) in trade receivable | 18 | ( | ( | ( | ||||||||||
| (Increase) in other receivables | 19 | ( | ( | ( | ||||||||||
| (Decrease)/increase in trade payables | ( | |||||||||||||
| Increase in other current liabilities | 20 | |||||||||||||
| Interest received | ||||||||||||||
| Interest paid | ( | ( | ( | |||||||||||
| Other finance income | ||||||||||||||
| Tax received/(paid) | 9 | ( | ( | |||||||||||
| Cash flow used in operating activities | $ | ( | $ | ( | $ | ( | ||||||||
| Investing activities | ||||||||||||||
| Purchase of intangible assets | 12 | ( | ( | ( | ||||||||||
| Purchase of property, plant and equipment | 13 | ( | ( | ( | ||||||||||
| Proceeds from sale of property, plant and equipment | 13 | |||||||||||||
| Acquisition of subsidiaries, net of cash acquired | 11 | |||||||||||||
| Investments in other non-current financial assets | 16 | ( | ( | ( | ||||||||||
Repayment of other non-current financial assets | 16 | |||||||||||||
| Cash flow used in investing activities | $ | ( | $ | ( | $ | ( | ||||||||
| Financing activities | ||||||||||||||
| Proceeds from issue of share capital | 20 | |||||||||||||
| Share issue costs | 20 | ( | ( | |||||||||||
| Proceeds from interest-bearing loans and borrowings | 15 | |||||||||||||
| Repayment of interest-bearing loans and borrowings | 15 | ( | ||||||||||||
| Payment of principal portion of lease liability | 14 | ( | ( | ( | ||||||||||
| Cash flow (used in)/from financing activities | $ | $ | ( | $ | ||||||||||
| Net cash flow during the period | ( | |||||||||||||
| Cash at bank and in hand at the beginning of the Period | ||||||||||||||
| Net foreign exchange difference | ( | ( | ||||||||||||
| Cash at bank and in hand at the end of the period | $ | $ | $ | |||||||||||
The accompanying notes are an integral part of the Consolidated Financial Statements.
F-10
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
1.General information
On January 27, 2021, Knilo HoldCo AB was registered as a Swedish public limited company and renamed as Olink Holding AB (publ) (the "Company" or "Group"). The Company was incorporated under the laws of Sweden as a limited company (“Aktiebolag”) and has its registered office in Uppsala, Sweden. The Company was incorporated on January 4, 2019 for the purpose of the acquisition of Olink OldCo AB (f/k/a Olink Proteomics Holding AB, “Olink Holdings”) and its subsidiaries. The Company business address is Salagatan 16F, SE-753 30, Uppsala, Sweden.
The Group develop, produce, market and sell biotechnological products and services along with thereof related activities. The Company has twelve wholly owned subsidiaries. On March 29, 2021, the Company completed its initial public offering (the "Offering") in the United States. The Company's American Depositary Shares ("ADSs") were approved for listing on The Nasdaq Global Market ("Nasdaq") under the trading ticker symbol "OLK". Trading on Nasdaq commenced at market open on March 25, 2021. The ultimate parent of the Company is Summa Equity Holding AB, Stockholm, Sweden.
The Company provides management services to its subsidiaries. The Company owns 100 % of Olink Finance AB, a company incorporated on 4 January 2019 under the laws of Sweden and has its registered office in Uppsala, Sweden. Olink Finance AB was used to acquire Olink Holdings on March 7, 2019 (“Olink Acquisition”).
The ultimate parent of the Company is Summa Equity Holding AB, Stockholm, Sweden. When referring to the Company and its subsidiaries collectively, they are referred to herein as the “Group”.
The Company's financial statements were authorized for issue by the Board of Directors on March 25, 2024.
2.Summary of Material Information about Accounting Policies
Material information about accounting policies applied in the preparation of these consolidated financial statements are set out below. These policies have been consistently applied to the consolidated financial statements for all periods presented, unless otherwise stated. Unless otherwise stated, all amounts are in thousands of U.S. Dollars.
2.1Basis of preparation
The consolidated financial statements of Olink Holding AB have been prepared in accordance with IFRS Accounting Standards ("IFRS"), as issued by the International Accounting Standards Board ("IASB"), and have been prepared using the historical cost measurement basis. There are no financial assets and liabilities measured at fair value on a recurring basis. The consolidated financial statements are presented in US dollars and all values are rounded to the nearest thousand ($000), except when otherwise indicated.
New and amended standards and interpretations
The following amendments will be applied for the first time in the annual reporting period commencing January 1, 2023:
•IFRS 17 Insurance contracts and Amendment to IFRS 17 Insurance contracts issued June 2020
•Amendments to IFRS 17 Insurance Contracts - Initial Application of IFRS 17 and IFRS 9 – Comparative Information
•Amendments to IAS 1 and IFRS Practice Statement 2 Disclosure of accounting policies
•Amendments to IAS 8: Accounting Policies, Changes in Accounting Estimates and Errors - Definition of Accounting Estimates,
•Amendments to IAS 12 Deferred tax related to assets and liabilities arising from a single transaction
•Amendments to IAS 12 International tax reform - Pillar two model rules
The amendments listed above did not have a material impact on the amounts recognized in the current period and are not expected to significantly affect future periods.
New and amended standards not yet effective
The following new accounting standards, amendments to accounting standards and interpretations have been published but are not mandatory for December 31, 2023 reporting periods and have not been early adopted by the Company.
•Amendments to IFRS 16: Lease liability measurement in sale and leaseback transaction - IFRS16,
•Amendments to IAS 1: Presentation of Financial Statements - Classification of Liabilities as Current or Non-current,
•Amendments to IAS 1: Amendments regarding the classification of debt with covenants
•Amendments to IAS 21 The Effects of Changes in Foreign Exchange Rates - Lack of exchangeability
F-11
•Amendments to IAS 7 and IFRS 7 Supplier finance arrangements
•Amendments to IFRS 10 and IAS 28: Sale or Contribution of Assets between an Investor and its Associate or Joint Venture
These standards, amendments or interpretations are not expected to have a material impact on the entity in the current or future reporting periods and on foreseeable future transactions.
2.2Basis of consolidation
The consolidated financial statements comprise the financial statements of the Company and its subsidiaries each period presented. Subsidiaries are all entities over which the Company has control. Control is achieved when the Company is exposed, or has rights, to variable returns from its involvement with the investee and has the ability to affect those returns through its power over the investee. Such subsidiaries are consolidated from the date on which control is transferred to the Group and are deconsolidated from the date that control ceases.
Assets, liabilities, income and expenses of a subsidiary acquired or disposed of during the period are included in the consolidated financial statements from the date the Group gain control until the date the Group ceases to control the subsidiary. A change in the ownership interest of a subsidiary, without a loss of control, is accounted for as an equity transaction.
All intra-group balances and transactions, and any unrealized income and expenses arising from intra-group transactions, are eliminated. Unrealized losses are eliminated in the same way as unrealized gains, but only to the extent that there is no evidence of impairment.
The accounting principles for subsidiaries have been changed, where applicable, to ensure a consistent application of the Group’s accounting principles.
2.3Material Information about Accounting Policies
i.Business combinations
Business combinations are accounted for using the acquisition accounting method. Identifiable assets acquired and liabilities assumed are measured initially at their fair values at the acquisition date. The excess of the consideration transferred, and the acquisition-date fair value of any previous equity interest in the acquiree, over the fair value of the identifiable net assets acquired is recognized as goodwill. The costs of effecting an acquisition are charged to the consolidated statement of income in the period in which they are incurred.
ii.Foreign currency translation
Functional and presentation currency
The consolidated financial statements are presented in U.S. Dollars. For the Parent and each subsidiary, the Group determines the functional currency and items included in the financial statements of each subsidiary are measured using that functional currency. In all cases the functional currency of a subsidiary is that of the primary country of operations of that subsidiary. On disposal of a foreign operation, the gain or loss that is reclassified to profit or loss reflects the amount that arises.
Transactions and balances
Foreign currency transactions of the Group are translated into the functional currency using the exchange rates prevailing on the transaction dates.
Monetary assets and liabilities denominated in foreign currencies are translated at the functional currency spot rates of exchange at the reporting date. Non-monetary assets and liabilities measured in terms of historic cost in a foreign currency are translated into the functional currency using the exchange rates prevailing on the initial transaction dates. Non-monetary items measured at fair value in a foreign currency are translated using the exchange rates prevailing on the date when the fair value is determined.
Differences arising on settlement or translation of monetary items excluding cash at bank in hand are recognized in other operating income/expense, and cash at bank in hand are recognized in foreign exchange gain in the consolidated statements of income.
Translation of foreign subsidiaries
F-11
The results and the financial position for the Parent and all the Group subsidiaries with a functional currency other than the U.S.Dollar are translated into U.S. Dollars as follows:
•Assets and liabilities at each balance sheet date are translated using the exchange rates prevailing at that balance sheet date;
•Income statements are translated using the average exchange rate prevailing at the corresponding month;
•Exchange differences arising on translation for consolidation are recognized in Other Comprehensive Income (“OCI”). On disposal of a foreign operation, the component of OCI relating to that particular foreign operation is reclassified to profit or loss; and
•Goodwill and fair value adjustments arising from the acquisition of foreign operations are treated as assets and liabilities in these operations and are translated to the exchange rate at the balance sheet date.
iii.Revenue recognition
The Group generates revenue from the sale of its products in the form of kits, provision of analysis services, and also from provision of custom development services. Value added tax and other sales taxes are excluded from revenue and products are generally sold without the right of return or rebates.
The Group accounts for a contract when the following criteria are met: the parties to the contract have approved the contract in which their rights, their obligations and the payment terms have been identified, the contract has commercial substance, and the collectability of the consideration is probable. Contracts with customers do not contain variable consideration.
Kit
Our Kit segment includes product sales of Explore, Flex, Focus and Target. The majority of the contracts for Kit products relate to sales orders containing single bundled performance obligations. Revenue from the sale of kits is recognized at the point in time when control of the products has transferred to the customer according to the shipping terms, typically Free Carrier (FCA) Incoterms. The average time from order to delivery is less than 1 month.
Analysis Services
The Group generates analysis services revenue from performing assay on customer samples to generate data on protein biomarkers. Revenue from the services is recognized at the point in time that the results of the analysis are transferred electronically to the customer as the customer does not control the asset created, and Olink does not have a right to payment until delivery of the results of the analysis.The majority of the analysis services contracts relate to sales orders containing single bundled performance obligation for the performance of services at fixed prices. Analysis services are sold at a fixed price per sample without any volume discounts, rebates, or refunds. The average duration of services contracts is less than 2 months.
Custom development services
The Group generates custom development revenue from providing customer specified Kits not available in our standard product set. Custom development projects are quoted at fixed price and extend over several months. Revenue from the development of the plates, a manufactured component in our kits, of custom development services is recognized over time since the Group has no alternative use for the asset created and has an enforceable right to payment for performance completed to date. These contracts contain a single bundled performance obligation being the provision of custom development services of panels for performing assays on samples. The Group uses an input method to determine the progress completed of custom development service arrangements because there is a direct relationship between the effort (i.e. based on costs incurred against expected total costs) and the transfer of service to the customer.
The average duration of a custom development service contract is less than 12 months.
iv.Cost of revenue
Cost of revenue primarily consists of manufacturing costs incurred in the production process including personnel and related costs; costs of component materials; depreciation from property, plant, and equipment; manufacturing overhead; delivery costs and allocated facilities and information technology related costs. In addition, cost of revenue includes royalty costs for licensed technologies included in our products, and write downs to net realizable value for slow-moving and obsolete inventory.
v.Leases
F-12
When the Group enters into contractual agreements, an assessment is performed to determine if the contract contains a lease. The Group identifies a lease if it conveys the right to control the use of an identified asset for a specific period in exchange for a determined consideration. At inception, a right-of-use asset for the underlying asset and corresponding lease liability are presented in the consolidated balance sheet measured on a present value basis except for short-term leases (expected term of 12 months or less) and leases with low value underlying asset for which payments are recorded as an expense on a straight-line basis over the lease term.
Right-of-use assets are measured at cost, less any accumulated depreciation and impairment losses, and adjusted for any remeasurement of lease liabilities. The right-of-use-assets are depreciated on a straight-line basis over the expected lease term of the underlying asset. Non-lease components are accounted for separately from the lease components.
Lease liabilities are measured at present value of non-cancellable payments of the expected lease term, which are mostly made of fixed payments of rent excluding maintenance fees; variable payments that are based on an index or a rate; amounts expected to be payable as residual value guaranties and extension or termination option if reasonably certain to be exercised.
The Group estimates the lease term in order to calculate the value of the lease liability at the initial date of the lease. Management uses judgement to determine the appropriate lease term based on the conditions of each lease. The Group considers all facts that create incentive to exercise an extension option or not to take a termination option including leasehold improvements, significant modification of the underlying asset or a business decision. The extension or termination options are only included in the lease term if it is reasonably certain of being exercised.
The discount rate used in the present value calculation is the incremental borrowing rate ("IBR") unless the implicit interest rate in the lease can be readily determined. The Group estimates the incremental borrowing rate for each lease or portfolio of leased assets, as most of the implicit interest rates in the leases are not readily determinable. To calculate the incremental borrowing rate, the Group considers its credit worthiness, the term of the arrangement, any collateral received and the economic environment. The incremental borrowing rates are subject to change mainly due to changes in the economic environment.
The lease liabilities are subsequently adjusted to reflect interest on the lease liabilities and lease payments made. Lease liabilities are remeasured (along with the corresponding adjustment to the right-of-use asset), whenever the following situations occur; a modification in the lease term, a change in the assessment of an option to purchase, a modification in the residual guarantees or in future lease payments due to a change of an index or rate tied to the payments. In addition, upon partial or full termination of a lease, the difference between the carrying amounts of the lease liability and the right-of-use asset is recorded in the consolidated statements of earnings.
vi.Goodwill
Goodwill arises on business combinations and is the excess of the consideration transferred over the net identifiable assets acquired and liabilities assumed. Goodwill is stated at cost less impairments, is deemed to have an indefinite useful life and is tested for impairment at least annually.
vii.Intangible assets
Intangible assets are stated at cost less provisions for amortization and impairments. Intangible assets acquired separately are measured on initial recognition at cost. The cost of intangible assets acquired in a business combination is their fair value at the date of acquisition.
Licenses separately acquired or acquired as part of a business combination are amortized over their estimated useful lives, using the straight-line basis, from the time they are available for use.
Customer relationships and technology acquired as part of a business combination are amortized over their estimated useful lives, using the straight-line basis.
Brands acquired as part of a business combination are deemed to have indefinite useful lives. The acquired brands are well-established within the industry, as evidenced by continued demand from and collaboration with blue chip research institutions. Further, the business is expected to operate under these brands for the foreseeable future, thus supporting the indefinite classification. These intangible assets are not amortized, but are tested for impairment annually, at the cash- generating unit level. The assessment of indefinite life is reviewed annually for the two cash generating units ("CGU's"), Analysis Services and Kits, to determine whether the indefinite life continues to be supportable. If not, the change in useful life from indefinite to finite is made on a prospective basis.
Licenses and customer relationships have estimated useful lives of up to 10 years and research and development technology have estimated useful lives of up to 15 years. Asset lives are reviewed, and where appropriate adjusted, annually.
F-13
Research and Development costs
Expenditure on research activities is recognized in the consolidated statement of income as incurred. Development expenditure from a project is capitalized as an intangible asset only if the expenditure can be measured reliably, the product or process is technically and commercially feasible, future economic benefits are probable and the Group intends to and have sufficient resources to complete development and to use or sell the asset. Otherwise, it is recognized in consolidated statement of income as research and development expenses. Subsequent to initial recognition, development expenditure is measured at cost less accumulated amortization and any accumulated impairment losses.
Amortization begins from the time the asset is ready for use. Depreciation is made on a straight-line basis over the useful life. The useful life is determined when the development project is finished and is estimated to 5 years.
viii.Property, plant and equipment
Property, plant and equipment ("PP&E") includes buildings; leasehold improvements; plant and machinery; furniture fittings and equipment; and assets under construction. PP&E is stated at the cost of purchase or construction, less depreciation and impairment. Depreciation is calculated to write off the cost less residual value of PP&E, excluding freehold land, using the straight-line basis over the expected useful life. Residual values and lives are reviewed, and where appropriate adjusted annually. The normal expected useful lives of the major categories of PP&E are:
•Leasehold improvements | shorter of the lease term or useful life | ||||
•Plant and machinery | |||||
•Furniture, fittings and equipment | |||||
On disposal of PP&E, the cost and related accumulated depreciation and impairments are removed from the balance sheet and the net amount, less any proceeds, is recognized in the income statement.
ix.Impairment of non-current assets
The carrying values of non-current assets are reviewed for impairment, either on a stand-alone basis or as part of a larger CGU, when events or changes in circumstances indicate the carrying value may be impaired. The Company assesses at each reporting date whether any such events or changes in circumstances exist. Additionally, goodwill, intangible assets with indefinite useful lives and intangible assets which are not yet available for use are tested for impairment annually. An impairment loss is recognized for the amount by which the asset’s carrying amount exceeds its recoverable amount. The recoverable amount is the higher of an asset’s fair value less costs of disposal and value in use. For the purposes of assessing impairment, assets are grouped at the lowest levels for which there are largely independent cash inflows, for Olink this is Analysis Services and Kits. Any provision for impairment is charged to the consolidated statement of income in expense categories consistent with the function of the impaired asset.
Impairments of goodwill are not reversed. Impairment losses on other non-current assets are only reversed if there has been a change in estimates used to determine recoverable amounts and only to the extent that the revised recoverable amounts do not exceed the carrying values that would have existed, net of depreciation or amortization, had no impairments been recognized.
x.Inventories
Inventory is stated at the lower of cost or net realizable value on a first-in, first-out basis. Cost comprises direct materials, direct labour and an appropriate proportion of variable and fixed overhead expenditure, the latter being allocated based on normal operating capacity. Cost is determined using a standard cost system, whereby the standard costs are updated periodically to reflect current costs.
The Group estimates the recoverability of inventory by referencing estimates of future demands and product life cycles, including expiration. The Group periodically analyses its inventory levels to identify inventory that may expire prior to expected usage or no longer meets quality specifications. Raw materials and finished goods within 180 days of expiration are considered slow moving and are considered obsolete when they are past the expiration date. When we have identified inventories to be in excess or obsolete, we write down the value of those inventories to their net realizable value based upon judgment and estimates about future demand and market conditions.
The Group develops new antibodies to be added to our Kit products. For these antibodies, the outcome of the process is uncertain due to the nature of the materials and process for creating them. The Group determines the cost of inventory using the historical success rate, updated regularly, based on internal testing of technical feasibility and expenses the cost
F-14
to research and development expenses in the consolidated statement of income for the portion expected to not be successful. Direct and indirect manufacturing costs incurred during research and development activities are expensed to research and development expense in the consolidated statement of income as consumed.
xi.Financial instruments
Financial assets
The Group’s financial assets are comprised of cash and cash equivalents, trade and other receivables, restricted cash and other non-current assets. All financial assets are recognized initially at fair value. Purchases and sales of financial assets are recognized on the settlement date, being the date upon which the Group commits to purchase or sell the asset.
Trade receivables
Trade receivables are measured at amortized cost and are carried at the original invoice amount less ECL allowance. For trade receivables, the Group applies a simplified approach in calculating ECLs. Therefore, the Group does not track changes in credit risk, but instead recognizes a loss allowance based on lifetime ECLs at each reporting date. The Group has established a provision matrix that is based on its historical credit loss experience.
When a trade receivable is determined to have no reasonable expectation of recovery it is written off against any ECL allowance available and then to selling expenses in the consolidated statement of income. Subsequent recoveries of amounts previously provided for or written off are credited selling expenses.
Cash at bank and in hand and restricted cash
Cash at bank and in hand are measured at amortized cost and includes cash on hand and call deposits with financial institutions.
Restricted cash includes deposits with contractual restrictions, fully restricting access to the principal amount. Only interest income from these deposits is available for use. Classification as current or non-current depends on the remaining restriction period. Refer to Note 16 for amounts and classification.
Financial liabilities
The Group’s financial liabilities include trade and other payables, loans and borrowings, and other liabilities. All financial liabilities, except lease liabilities, are recognized initially at fair value.
Financial liabilities are classified, at initial recognition, as financial liabilities at FVTPL, loans and borrowings, payables, or as derivatives designated as hedging instruments in an effective hedge, as appropriate. All financial liabilities are recognized initially at fair value and, in the case of loans, borrowings and payables, net of directly attributable transaction costs.
Loans and borrowings are subsequently carried at amortized cost, with the difference between the proceeds, net of transaction costs, and the amount due on redemption being recognized as a charge to the consolidated statements of other comprehensive income over the period of the relevant borrowing.
Derecognition of financial assets and liabilities
Financial assets are derecognized when the contractual rights to the cash flows from the financial asset expire or transfer and the Group has transferred substantially all the risks and rewards of ownership. Financial liabilities are derecognized when the contractual obligations are discharged or cancelled or expired. When the terms of a financial liability are modified, and not derecognized, the gain or loss is recognized in the statement of income and other comprehensive income. The gain or loss is the difference between the original contractual cash flows and the modified cash flows discounted to the original effective interest rate.
xii.Share-based payments
The Group operates restricted stock unit awards (RSU) and equity-settled stock option plans (ISO) under which the Group receives services from employees, officers and directors as consideration for equity instruments. Information relating to these schemes is set out in Note 21.
The fair value of the share-based payments is established on the closing price of ADS's of the Company on NASDAQ for the RSU's at grant date and using the Black-Scholes option pricing model for the ISO's. The number of RSU's and ISO's expected to vest are estimated on the grant date and subsequently revised on each reporting date.
F-15
Stock options
The fair value of options granted under the stock options program, adjusted for expectations related to forfeitures, is recognized as an employee benefits expense in selling, administration, and research and development expenses in the consolidated statement of income, with a corresponding increase in other contributed capital over the vesting period.
The total expense is recognized over the vesting period of four years , which is the period over which the vesting conditions are to be satisfied. At the end of each period, the Company revises its estimates of the number of options that are expected to vest based on the service conditions. It recognizes the impact of the revision to original estimates, if any, in profit or loss, with a corresponding adjustment to contributed capital.
Restricted stock units
Under the employee restricted stock units scheme, the RSU’s will vest during a four-year period and new shares will be issued when the RSU vest for no cash consideration. Over the vesting period, the market value of the RSU’s is recognized as an employee benefits expense in selling, administration, research and development, and cost of revenue in the consolidated statement of income, with a corresponding increase in contributed capital.
The total expense is recognized over the vesting period of four years , which is the period over which the vesting conditions are to be satisfied. At the end of each period, the Company revises its estimates of the number of RSU's that are expected to vest based on the service conditions. It recognizes the impact of the revision to original estimates, if any, in profit or loss, with a corresponding adjustment to contributed capital.
xiii.Current and deferred income tax
Income taxes are accounted for using the liability method of accounting. Current income tax is provided at the amounts expected to be paid, applying tax rates that have been enacted or substantively enacted by the balance sheet date.
Deferred tax assets and liabilities are determined based on deductible or taxable temporary differences between the amounts reported for consolidated financial statement purposes and the tax values of the assets and liabilities using enacted or substantively enacted tax rates that will be in effect for the year in which the differences are expected to be recovered or settled. Deferred tax assets and liabilities are recognized in earnings, in other comprehensive income or in equity based on the classification of the item to which they relate. Temporary differences attributable to the following assets and liabilities are not provided for:
•the initial recognition of goodwill,
•the initial recognition (other than in business combinations) of assets or liabilities that affect neither accounting nor taxable profit,
•differences related to investments in subsidiaries, associated companies and joint ventures to the extent that they will probably not reverse in the foreseeable future, and for which the Company is able to control the timing of the reversal of the temporary differences.
A deferred tax asset is recognized only to the extent that it is probable that future taxable profits will be available against which the asset can be utilized. In the calculation of deferred taxes, enacted or substantively enacted tax rates are used for the individual tax jurisdictions.
F-16
3.Significant Accounting estimates and judgements
In preparing these consolidated financial statements, management is required to make significant judgments and estimates that affect the reported amounts of revenues, expenses, assets, liabilities, and equity in the consolidated financial statements and the accompanying disclosures. Estimates and judgements are continuously evaluated and are based on historical experience and other factors, including expectations of future events.
In the process of applying the Group’ accounting policies, management has made the following judgements and estimates, which have the most significant effect on the amounts recognized in the consolidated Group financial statements:
Impact of ongoing conflicts
We are continuing to closely monitor how the armed conflict between Russia and Ukraine, as well as the Israel-Hamas conflict, are affecting our business. As of December 31, 2023, we concluded there was no evidence of material changes to recoverability risk of business assets, including deferred tax assets and trade receivables. Olink does not have significant sales or direct supply from Russia, Belarus, Ukraine, or the proximate regions affected by the Israel-Hamas conflict, though the impact from the armed conflicts on macro-economic conditions is currently unknown and could in the future have a negative effect on our results of operations, cash flows, financial condition or growth plans.
We continue to closely monitor our IT systems based on the general risk of potential cyberattacks by state or quasi-state actors as a result of the conflict between Russia and Ukraine.
We continue to closely monitor our IT systems based on the general risk of potential cyberattacks by state or quasi-state actors as a result of the conflict between Russia and Ukraine.
3.1.Share-based compensation
Key sources of estimation uncertainty
The Group estimates the cost of equity-settled transactions with employees and non-employees by reference to the fair value of the equity instruments at the date at which they are granted. The assumptions and models used for estimating the fair value of share-based compensation transactions are disclosed in Note 21. The Group also estimates a forfeiture rate to calculate the stock-based compensation expense for the awards. The forfeiture rate is estimated based on an analysis of actual forfeitures.
3.2. Impairment of Goodwill and Indefinite Lived Asset
Key sources of estimation uncertainty
In accordance with the accounting policy described in "ix" in Note 2.3, the Group annually performs an impairment test on goodwill and indefinite lived assets. The recoverable amount of each CGU has been determined based on its value in use calculation, which includes estimates about their future financial performance based on cash flows approved by management.
Accounting Judgement
Asset impairment requires managements judgement, particularly in assessing factors such as our ability to continue developing and expanding products and services offered to address emerging business demands, and our ability to hire and retain qualified professionals can affect future cash flows, and actual results might differ from future cash flows used in the goodwill impairment test. The assumptions used for estimating fair value and assessing available headroom based on conditions that existed at the testing date are disclosed in note 12. Historically, the Company has not recorded an impairment charge on goodwill.
3.3.Deferred Taxes
Accounting Judgement
The Group has recognized deferred tax assets for fiscal loss carry-forwards, and deductible temporary differences. The Group considers the analysis of forecast and future tax planning strategies. Estimates of taxable profit are made based on the forecast which are aligned with goodwill impairment testing assumptions, on an undiscounted basis. Management judgement is required concerning uncertainties that exist with respect to the timing of future taxable income required to recognize a deferred tax asset. At period end, we assess whether there is convincing evidence that the Group will generate future taxable income against which deferred tax assets can be utilized and, thus, that recovery is probable. See Note 9.
3.4Leases
Key sources of estimation uncertainty
The discount rate is used to determine the initial carrying amount of the lease liabilities and the right-of-use assets. The Group cannot readily determine the interest rate implicit in the lease, therefore, it uses its IBR to measure lease liabilities. The Group estimates the IBR using observable inputs (such as market interest rates) when available and is required to make certain entity-specific estimates.
Accounting Judgement
F-17
At initial recognition and subsequent remeasurement, management uses judgement to determine the appropriate term applied in a lease contract. The outcome may turn out not to match the actual outcome of the lease and may have an adverse effect on the right-of-use assets. In determining the lease term, management considers all facts and circumstances that create an economic incentive to exercise an extension option, or not exercise a termination option. Extension options (or periods after termination options) are only included in the lease term if the lease is reasonably certain to be extended (or not terminated).
3.5Development costs
Accounting Judgement
The Group has a process to determine whether development costs meet the criteria for capitalization. However, based on management’s judgement and the nature of the development activities, such criteria and in particular technical and economic feasibility is normally not met until the development phase is complete.
F-18
4.Financial risk management
Financial risk factors
The Group's activities are subject to several financial risks: market risk (including currency risk and interest rate risk), credit risk and liquidity risk. The Group strive to minimize potential unfavorable effects from these risks on the Group´s financial results.
The aim of the Group´s financial operations is to:
•Ensure that the Group can meet their financial obligations timely,
•Manage financial risks, and
•Ensure a supply of necessary financing.
The Group´s risk management is predominantly controlled by senior management.
Market risk - Currency risk (transaction risk)
The Group operates internationally and are exposed to currency risk where invoicing is made in a currency other than the functional currency of the relevant Group entity. Primarily, the Group is exposed to currency risk in Group companies with SEK as the functional currency. The primary risks in these companies are USD/SEK, EUR/SEK, GBP/SEK and JPY/SEK due to sales (trade receivables and royalties) and purchases (trade payables and accrued expenses). Mitigation of this risk occurs naturally by matching expenses and obtaining borrowings, as required, in the same foreign currency. The currency risk is monitored on a regular basis. The Group has not entered into derivative currency instruments during the reported period.
Exposure
The Group's primary exposure to foreign currency risk at the end of the reporting period was as follows:
| As of December 31, 2023 | ||||||||||||||
| Amounts in thousands of USD | U.S.$ | EUR | GBP | JPY | ||||||||||
| Trade receivables | $ | $ | $ | $ | ||||||||||
| Trade payable | ||||||||||||||
| Royalties | ||||||||||||||
| Accrued expenses | ||||||||||||||
| As of December 31, 2022 | ||||||||||||||
| Amounts in thousands of USD | U.S.$ | EUR | GBP | JPY | ||||||||||
| Trade receivables | $ | $ | $ | $ | ||||||||||
| Trade payable | ||||||||||||||
| Royalties | ||||||||||||||
| Accrued expenses | ||||||||||||||
Sensitivity
The following table demonstrates the sensitivity to a reasonably possible change in USD, EUR, GBP and JPY against SEK as of December 31, 2023 and 2022, with all other variables held constant. The impact on the Group’s loss before tax is due to changes in the fair value of monetary item assets and monetary liabilities. There is no additional impact on the components of equity because the Group did not have any item that directly affected equity. The Group’s exposure to foreign currency changes for all other currencies than SEK is not material.
The below analysis is based on a foreign currency rate changes of 3 % on the USD, EUR, GBP and JPY.
The Group's risk exposure in foreign currencies:
F-19
| As of December 31, 2023 | |||||
| Impact of non-functional currency foreign exchange exposures (Amounts in thousands of USD) | (Increase)/decrease in loss before tax | ||||
USD/SEK exchange rate - increase 3% | ( | ||||
USD/SEK exchange rate - decrease 3% | |||||
EUR/SEK exchange rate - increase 3% | |||||
EUR/SEK exchange rate - decrease 3% | ( | ||||
GBP/SEK exchange rate - increase 3% | |||||
GBP/SEK exchange rate - decrease 3% | ( | ||||
JPY/SEK exchange rate - increase 3% | |||||
JPY/SEK exchange rate - decrease 3% | ( | ||||
| As of December 31, 2022 | |||||
| Impact of non-functional currency foreign exchange exposures (Amounts in thousands of USD) | (Increase)/decrease in loss before tax | ||||
USD/SEK exchange rate - increase 3% | |||||
USD/SEK exchange rate - decrease 3% | ( | ||||
EUR/SEK exchange rate - increase 3% | |||||
EUR/SEK exchange rate - decrease 3% | ( | ||||
GBP/SEK exchange rate - increase 3% | |||||
GBP/SEK exchange rate - decrease 3% | ( | ||||
JPY/SEK exchange rate - increase 3% | |||||
JPY/SEK exchange rate - decrease 3% | ( | ||||
Market risk - Interest-rate risk
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates.
As of December 31, 2023, the Group does not have any outstanding debt or other debt structures other than leasing. The Group does not hold any fixed-income investments.
Interest rate derivative instruments were not used by the Group during the reported periods.
Credit risk
Credit risk is the risk that a counterparty will not meet its obligations under a financial instrument or customer contract, leading to a financial loss. The Group is exposed to credit risk from its operating activities (primarily trade receivables) and from its investing activities, including deposits with banks and financial institutions and foreign exchange transactions. Credit risk relates primarily to customer credit limits, which are subject to certain credit rating rules and authorization processes. However, the majority of the Group customer base tend to be blue chip global companies and therefore such customers usually have strong credit ratings. Group’s sales are concentrated such that 43 % of sales in 2023 and 45 % of sales in 2022 are with customers based in the U.S.
The maximum default risk for the Group is equivalent to the net receivables reported in the Consolidated Financial Statements. The Group have historically almost non-existent credit losses and based on historical data together with a forward-looking assessment, the 2023 expected credit loss for trade receivables is disclosed in Note 18, ‘Trade receivables’.
The Group´s cash at bank is held in Investment Grade credit rated banks. To mitigate the counterparty risk cash is distributed among different banks and it is monitored on a regular basis.
Other financial assets at amortized cost include rental deposits. The credit risk for other financial assets at amortized cost as at December 31, 2023 and 2022 is not material and no credit loss reserve has been recognized.
F-20
Liquidity risk
Cash at bank allows the Group to meet its liquidity risk obligations as they come due. Following the Initial Public Offering of the Group in March, 2021 the liquidity risk has been managed by cash at bank deposits.
The following table includes an analysis of the Company’s financial liabilities, grouped according to their maturity dates based on contractual undiscounted payments and considers the period remaining until their contractual maturity date as at December 31, 2023 and 2022:
| amounts in thousands USD | |||||||||||||||||
| As per December 31, 2023 | Total | Less than 1 year | 1 to 3 years | 3 to 5 years | More than 5 years | ||||||||||||
| Lease liabilities (Note 15.1) | |||||||||||||||||
| Accounts payable (Note 15.2) | — | — | — | ||||||||||||||
| Salaries and wages (Note 15.2) | — | — | — | ||||||||||||||
| Royalties (Note 15.2) | — | — | — | ||||||||||||||
| Accrued expenses (Note 15.2) | — | — | — | ||||||||||||||
| As per December 31, 2022 | Total | Less than 1 year | 1 to 3 years | 3 to 5 years | More than 5 years | ||||||||||||
| Lease liabilities (Note 15.1) | |||||||||||||||||
| Accounts payable (Note 15.2) | — | — | — | ||||||||||||||
| Salaries and wages (Note 15.2) | — | — | — | ||||||||||||||
| Royalties (Note 15.2) | — | — | — | ||||||||||||||
| Accrued expenses (Note 15.2) | — | — | — | ||||||||||||||
4.2Capital management
For the purpose of the Group's capital management, capital includes issued capital, other contributed capital and all other equity reserves attributable to the equity holders of the Company. The Group's capital structure and dividend policy is decided by the board of directors, The Financial operations continuously reviews the Group's capital structure considering amongst other things, market conditions, financial flexibility, business risk, and growth rate. The primary objective of the Group's’ capital management is to maximize the shareholder value.
5.Segment and revenue information
5.1Description of segments and principal activities
Operating segments are reported based on the financial information provided to the Chief Executive Officer (“CEO”). The CEO is identified as the Chief Operating Decision Maker (“CODM”) of the Company. The CODM monitors the operating results of its operating segments separately for the purpose of making decisions about resource allocation and performance assessment. Evaluation of segment performance is primarily based on revenue growth. The CODM monitors the operating segments based on revenue growth and gross profit under two segments: Kit and Service. All other operating segments have been aggregated and are included within the "All other segments" heading.
The Group’s research and development activities, sales & administrative activities, financing (including finance costs, finance income and other income) and income taxes are managed on a corporate basis and are not allocated to operating segments.
Capital expenditure consists of additions of property, plant and equipment and intangible assets.
F-21
5.2Revenue and Gross Profit
The following tables presents the Company’s key financial information by segment:
| Amounts in thousands of USD | For the year ended December 31, 2023 | For the year ended December 31, 2022 | For the year ended December 31, 2021 | ||||||||
| Kit | |||||||||||
| Revenue from external customers | $ | $ | $ | ||||||||
| Cost of revenue | ( | ( | ( | ||||||||
| Gross profit | $ | $ | $ | ||||||||
| Service | |||||||||||
| Revenue from external customers | |||||||||||
| Cost of revenue | ( | ( | ( | ||||||||
| Gross profit | $ | $ | $ | ||||||||
| Total segments | |||||||||||
| Revenue from external customers | |||||||||||
| Cost of revenue | ( | ( | ( | ||||||||
| Gross profit | $ | $ | $ | ||||||||
| All other segments | |||||||||||
| Revenue from external customers | |||||||||||
| Cost of revenue | ( | ( | ( | ||||||||
| Gross profit | $ | $ | $ | ||||||||
| Consolidated | |||||||||||
| Revenue from external customers | |||||||||||
| Cost of revenue | ( | ( | ( | ||||||||
| Gross profit | $ | $ | $ | ||||||||
F-22
5.3Disaggregation of revenue from contracts with customers
The Group is domiciled in Sweden.
| for the year ended December 31, 2023 | Kit | Services | All other segments | Total | ||||||||||
amounts in thousands USD | ||||||||||||||
| Sweden | $ | $ | $ | $ | ||||||||||
| United States | ||||||||||||||
| Americas (excluding US) | ||||||||||||||
| EMEA (excluding Sweden) | ||||||||||||||
| China | ||||||||||||||
| Japan | ||||||||||||||
| Rest of world | ||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||
for the year ended December 31, 2022 | Kit | Services | All other segments | Total | ||||||||||
| amounts in thousands USD | ||||||||||||||
| Sweden | $ | $ | $ | $ | ||||||||||
| United States | ||||||||||||||
| Americas (excluding US) | ||||||||||||||
| EMEA (excluding Sweden) | ||||||||||||||
| China | ||||||||||||||
| Japan | ||||||||||||||
| Rest of world | ||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||
for the year ended December 31, 2021 | Kit | Services | All other segments | Total | ||||||||||
amounts in thousands USD | ||||||||||||||
| Sweden | $ | $ | $ | $ | ||||||||||
| United States | ||||||||||||||
| Americas (excluding US) | ||||||||||||||
| EMEA (excluding Sweden) | ||||||||||||||
| China | $ | |||||||||||||
| Japan | ||||||||||||||
| Rest of world | ||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||
There were no customers in the Group in 2023, 2022 or 2021 periods that individually exceeded 10% of total revenue.
F-23
5.4Non-current assets by geography
Sweden is regarded as being the Company’s country of domicile. Non-current assets, excluding financial instruments and deferred tax asset, are distributed by geography as follows:
| Amounts in thousands USD | As of December 31, | |||||||
| 2023 | 2022 | |||||||
| Sweden | $ | $ | ||||||
| Rest of World | ||||||||
| Total | $ | $ | ||||||
6.Operating expenses by nature
| Amounts in thousands USD | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||
| Included in costs of revenue | |||||||||||
| Cost of inventories recognized as an expense | $ | $ | $ | ||||||||
| Depreciation of tangible assets (Note 13, 14.2) | |||||||||||
| Amortization of intangible assets (Note 12) | |||||||||||
| Employee compensation (Note 7) | |||||||||||
| Included in selling expenses | |||||||||||
| Depreciation of tangible assets (Note 13, 14.2) | |||||||||||
| Amortization of intangible assets (Note 12) | |||||||||||
| Employee compensation (Note 7) | |||||||||||
| Included in administrative expenses | |||||||||||
| Depreciation of tangible assets (Note 13, 14.2) | |||||||||||
| Amortization of intangible assets (Note 12) | |||||||||||
| Employee compensation (Note 7) | |||||||||||
| Included in research and development expenses | |||||||||||
| Depreciation of tangible assets (Note 13, 14.2) | |||||||||||
| Amortization of intangible assets (Note 12) | |||||||||||
| Employee compensation (Note 7) | |||||||||||
7.Employee Compensation
The Group operate defined-contribution plans for the benefit of its employees. The Group has no further payment obligations once the contributions have been paid. The Group’ contributions to defined contribution plans are expensed as incurred.
| Amounts in thousands USD | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||
| Salaries and wages | $ | $ | $ | ||||||||
| Share-based payments | |||||||||||
| Social security costs | |||||||||||
| Pension costs - defined contribution plans | |||||||||||
| Total employee benefits cost | $ | $ | $ | ||||||||
For information about stock-based compensation, please see note 21.
F-24
8.Financial income and expenses
The following table shows a reconciliation of financial income and expense. Interest expense on loans and other borrowings during 2021 relates to the shareholder loan and loan facilities, and interest expense on lease liabilities relates to our property and equipment leases, described in note 15.
| Amounts in thousands USD | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||
| Interest income from cash at bank and in hand | $ | $ | $ | ||||||||
| Interest expense on loans and other borrowings | ( | ( | ( | ||||||||
| Interest expense on lease liabilities | ( | ( | ( | ||||||||
| Total interest income/(expense) | ( | ||||||||||
| Foreign exchange gain on cash at bank and in hand | |||||||||||
| Other financial income/(expenses) | ( | ||||||||||
| Financial items - net | $ | $ | $ | ( | |||||||
9.Income tax
Items reported for income taxes include a reasonable estimate of the impact of the material aspects of the Swedish tax rate reduction which was signed into law on June 14, 2018, on the deferred tax assets and liabilities. Swedish tax rules reduced the corporate income tax from 21.4 % to 20.6 % from January 1, 2021. The major components of income tax benefit (expense) for the periods ended December 31, 2023, 2022, 2021 are as follows:
| Amounts in thousands USD | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||
| Current tax: | |||||||||||
| Current tax on profit for the year | $ | ( | $ | ( | $ | ( | |||||
| Deferred income tax | |||||||||||
| (Decrease)/increase in deferred tax assets | |||||||||||
| Decrease/(increase) in deferred tax liabilities | |||||||||||
| Total deferred tax expense/(benefit) | |||||||||||
| Income tax (expense)/benefit | $ | $ | $ | ||||||||
A reconciliation between reported tax expense for each period and the theoretical tax expense that would arise when applying statutory tax rate in Sweden, 20.6
| Amounts in thousands USD | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||
| Loss before tax | $ | ( | $ | ( | $ | ( | |||||
| Income tax calculated according to tax rate in Sweden 20.6% | |||||||||||
| Tax effects from: | |||||||||||
| Non-deductible costs | ( | ( | ( | ||||||||
| Previously unrecognized tax losses used to reduce current tax expenses | |||||||||||
| Differences in overseas tax rates | ( | ( | |||||||||
| Adjustments in respect of income tax of previous years | ( | ( | |||||||||
| Other | ( | ||||||||||
| Income tax | $ | $ | $ | ||||||||
F-25
Deferred tax balances
Deferred tax assets and liabilities of the Company are shown in the table below:
| Deferred tax assets | Lease Liabilities | Tax losses | Other | Total | ||||||||||
| Amounts in thousands USD | ||||||||||||||
| Balance as of January 1, 2021 | $ | $ | $ | $ | ||||||||||
| Recognized in the statement of comprehensive income | ||||||||||||||
| Recognized in statement of Equity | ||||||||||||||
| Exchange differences | ( | ( | ( | |||||||||||
| Balance as of December 31, 2021 | $ | $ | $ | $ | ||||||||||
| Recognized in the statement of comprehensive income | ||||||||||||||
| Recognized in statement of Equity | ||||||||||||||
| Exchange differences | ( | ( | ( | ( | ||||||||||
| Balance as of December 31, 2022 | $ | $ | $ | $ | ||||||||||
| Recognized in the statement of comprehensive income | ||||||||||||||
| Recognized in statement of Equity | ||||||||||||||
| Exchange differences | ||||||||||||||
| Balance as of December 31, 2023 | $ | $ | $ | $ | ||||||||||
| Deferred tax liabilities | Deferred tax on untaxed reserves | Intangibles & Inventory Valuation | Other Temporary Differences | Total | ||||||||||
| amounts in thousands USD | ||||||||||||||
| Balance as of January 1, 2021 | $ | $ | $ | $ | ||||||||||
| Recognized in the statement of comprehensive income | ( | ( | ( | |||||||||||
| Net from deferred tax asset | — | — | ||||||||||||
| Exchange differences | ( | ( | ( | |||||||||||
| Balance as of December 31, 2021 | $ | $ | $ | $ | ||||||||||
| Recognized in the statement of comprehensive income | ( | ( | ||||||||||||
| Exchange differences | ( | ( | ( | |||||||||||
| Balance as of December 31, 2022 | $ | $ | $ | $ | ||||||||||
| Recognized in the statement of comprehensive income | ( | ( | ||||||||||||
| Exchange differences | ||||||||||||||
| Balance as of December 31, 2023 | $ | $ | $ | $ | ||||||||||
The Group has tax losses that arose in Sweden of $77,091 thousand (2022: $40,683 thousand) that are available indefinitely for offsetting against future taxable profits of the entities in which the losses arose. It also has tax losses related to interest expense deductions that arose in Sweden of $15,047 thousand (2022: $14,924 thousand) that are available for up to 6 years for offsetting against future taxable profits of the entities in which the deduction arose. The year on year movement on tax losses related to interest expense deductions is solely related to changes in foreign exchange.
Based on management's projections regarding future taxable profits, the Group has recognized deferred tax assets for the former but not for the latter because it is not currently probable that the entities in which the loss arose will be able to generate sufficient taxable profits before these entities taxable deduction offsets expire after 6 years. Furthermore, these taxable deductions are not available to other group entities where profits are expected to arise. In evaluating the probability of realizing the deferred tax assets, the Company considered all available positive and negative evidence of future taxable income, including past operating results and forecasted market growth and earnings. During 2023, a gross movement of $10,439 thousand (2022 $1,755 thousand) was recorded in the deferred tax asset with a net impact of $8,272 thousand (2022 $2,725 thousand) on the annual results. If the Company were able to recognize all unrecognized deferred tax assets, net profit would increase by $3,100 thousand (2022: $3,074 thousand).
F-26
10.Investments in subsidiaries
The Company had the following subsidiaries as per December 31, 2023 and 2022:
| Country of registration and operation | Share of common shares owned by the Company (%) | |||||||||||||
| Name | Principle Activities | 2023 | 2022 | |||||||||||
| Olink Finance AB | Cash management | Sweden | ||||||||||||
| Olink OldCo AB | Other operational activities | Sweden | ||||||||||||
| Olink Proteomics AB | Sales, production, and research & development | Sweden | ||||||||||||
| Agrisera AB | Production, and research & development | Sweden | ||||||||||||
| Olink Proteomics Inc. | Sales of services and distribution services | USA | ||||||||||||
| Olink Proteomics Ltd | Marketing coordination and sales services | UK | ||||||||||||
| Olink Proteomics B.V | Marketing coordination and sales services | Netherlands | ||||||||||||
| Olink Proteomics GmbH . | Marketing coordination and sales services | Germany | ||||||||||||
| Olink Proteomics KK | Marketing coordination and sales services | Japan | ||||||||||||
| Olink Biotech (Shanghai) Co., Ltd | Distribution, marketing coordination and sales services | China | ||||||||||||
| Olink Proteomics SAS | Marketing coordination and sales services | France | ||||||||||||
| Olink Proteomics SG Pte. Ltd. | Marketing coordination and sales services | Singapore | N/A | |||||||||||
11.Business combination
Acquisitions in 2023
Acquisitions in 2023
No acquisitions were made in 2023 or in the preceding two fiscal years.
F-27
12.Goodwill and other intangible assets
Changes in goodwill and other intangible assets for the Company periods are as follows:
| Amounts in thousands USD | Goodwill | Customer relation | Technology | Brand and Licenses | Development Cost | Total | ||||||||||||||
| Cost | ||||||||||||||||||||
| As of January 01, 2022 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Additions | — | — | — | |||||||||||||||||
| Translation differences | ( | ( | ( | ( | ( | ( | ||||||||||||||
| As of December 31, 2022 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Additions | — | — | — | |||||||||||||||||
| Reclassifications | — | — | — | |||||||||||||||||
| Translation differences | ||||||||||||||||||||
| As of December 31, 2023 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Amortization and impairment | ||||||||||||||||||||
| As of January 01, 2022 | $ | — | $ | $ | $ | $ | $ | |||||||||||||
| Amortization | — | |||||||||||||||||||
| Translation differences | — | ( | ( | ( | ( | ( | ||||||||||||||
| As of December 31, 2022 | $ | — | $ | $ | $ | $ | $ | |||||||||||||
| Amortization | — | |||||||||||||||||||
| Reclassifications | — | — | — | |||||||||||||||||
| Translation differences | — | |||||||||||||||||||
| As of December 31, 2023 | $ | — | $ | $ | $ | $ | $ | |||||||||||||
| Net Book Value | ||||||||||||||||||||
| As of December 31, 2023 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| As of December 31, 2022 | $ | $ | $ | $ | $ | $ | ||||||||||||||
F-28
12.1Test of goodwill and indefinite lived assets impairment
For impairment testing, goodwill acquired through business combinations and brands with indefinite useful lives are allocated to the Kit and Services CGUs, which are reportable segments.
| Amounts in thousands USD | |||||||||||
| As of December 31, 2023 | Kit | Services | Total | ||||||||
| Goodwill | $ | $ | $ | ||||||||
| Brands | |||||||||||
| As of December 31, 2022 | Kit | Services | Total | ||||||||
| Goodwill | $ | $ | $ | ||||||||
| Brands | |||||||||||
The recoverable amounts of the CGUs’ value-in-use calculation is based on cash flow projections from financial budgets approved by senior management covering a ten-year period. The forecast period exceeds 5 years since the market for Olink's products is a relatively new market and we expect strong growth over the next 10 years.
The discount rates used in 2023 and 2022 are based on the Company’s WACC of 14 % and 17 % respectively, as both CGUs have integrated operations across the business. The discount rate is adjusted where appropriate for specific segment, country and currency risks. The valuation methodology uses significant inputs which are not based on observable market data; therefore, this valuation technique is classified as level 3 in the fair value hierarchy.
F-29
Details relating to the discounted cash flow models used in the impairment tests of the Kit and Services CGUs are as follows:
| Valuation basis | Value in use | ||||||||||
| Key assumptions | •Sales growth rates •Profit margins •Terminal growth rate •Discount rate | ||||||||||
| Determination of assumptions | •Sales growth rates are internal forecasts based on both internal and external market information •Profit margins are internal forecasts based on both internal and external market information •Terminal growth rates based on management’s estimate of future long-term average growth rates •Discount rates based on the Company’s WACC, adjusted where appropriate. | ||||||||||
| Period of specific projected cash flows | |||||||||||
| Terminal growth rate and discount rate | Terminal growth rate | Discount rate 2023/2022 | |||||||||
| Kit and Services CGUs | |||||||||||
The Company performed its annual goodwill impairment test for each of its reporting units during the fourth quarter in 2023 and 2022 using a discounted cash flow analysis, concluded that the recoverable amounts of all of its reporting units were in excess of their carrying values. No impairment of goodwill was required.
The discounted cash flow analysis includes management’s current assumptions as to future cash flows and long-term growth rates. A sensitivity analysis including all key assumptions is performed and management believe that no reasonably possible change in any of the above key assumptions would cause the carrying value to materially exceed the recoverable value. For all cash generating units there is sufficient headroom before any changes in key assumptions would cause a valuation adjustment. The performed sensitivity analysis demonstrates that the value of goodwill and other intangible assets with indefinite useful life is more than defensible even if the discount rate is increased with one and a half percentage points and if the growth rate after the forecast period is decreased with two percentage points for all cash generating units. Even forecasts for sales growth and profit margins are included in the sensitivity analysis and no reasonable changes in these would cause a need of impairment.
F-30
13.Property, plant and equipment
Changes in property, plant and equipment for the Group are as follows:
| Amounts in thousands USD | Buildings | Leasehold improvement | Plant and machinery | Furniture, fittings and equipment | Construction in progress for property, plant and equipment | Total | ||||||||||||||
| Cost | ||||||||||||||||||||
| As of January 01, 2022 | $ | $ | $ | $ | $ | |||||||||||||||
| Additions | ||||||||||||||||||||
| Transfers | ( | ( | ||||||||||||||||||
| Disposals | ( | ( | ( | — | ( | |||||||||||||||
| Translation differences | ( | ( | ( | ( | ( | |||||||||||||||
| As of December 31, 2022 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Additions | ||||||||||||||||||||
| Transfers | ( | ( | ||||||||||||||||||
| Disposals | ( | ( | ( | — | ( | |||||||||||||||
| Translation differences | ||||||||||||||||||||
| As of December 31, 2023 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Amortization and impairment | ||||||||||||||||||||
| As of January 01, 2022 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Depreciation for the period | — | |||||||||||||||||||
| Transfers | ( | — | ||||||||||||||||||
| Disposals | ( | ( | — | ( | ||||||||||||||||
| Translation differences | ( | ( | ( | — | ( | |||||||||||||||
| As of December 31, 2022 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Depreciation for the period | — | |||||||||||||||||||
| Transfers | ( | ( | — | ( | ||||||||||||||||
| Disposals | ( | — | ( | |||||||||||||||||
| Translation differences | — | |||||||||||||||||||
| As of December 31, 2023 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| Net Book Value | ||||||||||||||||||||
| As of December 31, 2023 | $ | $ | $ | $ | $ | $ | ||||||||||||||
| As of December 31, 2022 | $ | $ | $ | $ | $ | $ | ||||||||||||||
14.Leases
The Group is a lessee
The Group have lease contracts for various items of property and production equipment used in its operations. Lease terms for properties and equipment are generally up to 10 years. Certain leases include extension and termination options. These options are negotiated by management to provide flexibility in managing the leased-asset portfolio and align with the Group's business needs. The Group has not signed any material contracts that have not yet started as of December 31, 2023.
For the year ended December 31, 2023 and 2022, the Group had lease contracts with lease terms of 12 months or less. The Group applied the ‘short term lease’ recognition for these leases. The Group had leases pertaining to assets of low value, such as office equipment. The Group applied the ‘lease of low-value assets’ recognition exemptions in IFRS 16 for these leases, meaning the value of these contracts is not part of the right-of-use asset or leases liability.
F-31
14.1Amounts recognized in the consolidated balance sheet
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
| Right-of-use assets | ||||||||
| Property | $ | $ | ||||||
| Equipment | ||||||||
| Total assets | $ | $ | ||||||
| Lease liabilities | ||||||||
| Current (Note 15.1) | ||||||||
| Non-current (Note 15.1) | ||||||||
| Total liabilities | $ | $ | ||||||
The additions of right-of-use assets during the Group periods ended December 31, 2023 and 2022 were $20,145 thousand and $4,908 thousand, respectively.
14.2Amounts recognized in the consolidated statement of income related to leases
| Amounts in thousands USD | for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | ||||||||
| Depreciation charge of right-of-use assets | |||||||||||
| Property | $ | $ | $ | ||||||||
| Equipment | |||||||||||
| Total depreciation of right-of-use-assets | |||||||||||
| Interest expense (included in finance cost, Note 8) | |||||||||||
| Total amount recognized in net loss for the period | $ | $ | $ | ||||||||
No significant variable lease payments that are not included in the lease liability have been identified for the Group. Short term lease payments and payments on low value lease assets were $233 thousand for the year ended December 31, 2023 and $436 thousand for the year ended December 31, 2022.
The total cash outflow for leases during the periods ended December 31, 2023 and 2022 were $2,683 thousand and $2,908 thousand, respectively. The maturity analysis of lease liabilities for the Company is disclosed in Note 4.1.
15.Financial instruments per category
The following tables present the Group’s financial instruments per category
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
| Current asset instruments at amortized cost | ||||||||
| Trade receivables | $ | $ | ||||||
| Total current asset instruments at amortized cost | ||||||||
| Non-current asset instruments at amortized cost | ||||||||
| Other non-current receivables | ||||||||
| Total non-current asset instruments at amortized cost | ||||||||
| Total financial assets | $ | $ | ||||||
F-32
15.1Financial liabilities: Interest-bearing loans and borrowings
| Amounts in thousands USD | Interest Rate | Maturity | As of December 31, 2023 | ||||||||
| Current interest-bearing loans and borrowings | |||||||||||
| Lease Liabilities (Note 14) | 2024 | $ | |||||||||
| Non-current interest-bearing loans and borrowings | |||||||||||
| Lease Liabilities (Note 14) | 2024-2033 | $ | |||||||||
| Total interest-bearing loans and borrowings | $ | ||||||||||
| Amounts in thousands USD | Interest Rate | Maturity | As of December 31, 2022 | ||||||||
| Current interest-bearing loans and borrowings | |||||||||||
| Lease Liabilities (Note 14) | 2023 | $ | |||||||||
| Non-current interest-bearing loans and borrowings | |||||||||||
| Lease Liabilities (Note 14) | 2023-2032 | $ | |||||||||
| Total interest-bearing loans and borrowings | $ | ||||||||||
Other interest-bearing loan facility
During the year ended December 31, 2019 the Group entered into a loan facility in the amount of $110 million with Bridgepoint Credit and DNB AB (Publ) as part of the financing of the Olink Acquisition (Facilities). During the year ended December 31, 2020 we amended our debt structure under the existing loan facility with Bridgepoint Credit and DNB AB (Publ), increasing the total commitment under the facilities to $137.6 million. The effective date of the amended agreement was December 23, 2020.
A total of $63.5 million had been drawn down under the term Facility B, adjusted for transaction costs of $1.8 million. The loans were raised in USD and EUR to match revenue streams in USD and EUR. The remaining undrawn credit under the facilities was $74.1 million. Under the terms of the Facilities, the Group pledged the assets, including patents and other intellectual property, of its subsidiary, Olink Proteomics Inc.
On March 30, 2021, we repaid $65.6 million of outstanding loan facilities plus accrued interest of $1.9 million using the net proceeds from the offering and had no outstanding loan balances. As of December 31, 2023, we had $121.0 million in cash at bank, restricted cash of $1.4 million (see note 16) and no outstanding loan balances.
15.2Other financial liabilities
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
| Other financial liabilities at amortized cost | ||||||||
| Accounts payable | $ | $ | ||||||
| Salaries and wages | ||||||||
| Royalties | ||||||||
| Accrued expenses | ||||||||
| Total current financial liabilities | $ | $ | ||||||
F-33
15.3 Changes in Liabilities attributable to financing activities
The following tables show changes in liabilities attributable to financing activities for the Group respectively:
| Amounts in thousands USD | Current Interest bearing liabilities (excluding current lease liabilities) | Current lease liabilities | Non-current Interest bearing liabilities (excluding Non-current lease liabilities) | Non- current lease liabilities | Total liabilities from financing activities | ||||||||||||
| Liabilities as of January 1, 2021 | $ | $ | $ | $ | $ | ||||||||||||
| Cash flows | — | ( | ( | — | ( | ||||||||||||
| Non cash-flow: | |||||||||||||||||
| New leases | — | — | |||||||||||||||
| Foreign exchange adjustments | — | ( | ( | ||||||||||||||
| Other | ( | ||||||||||||||||
| Liabilities as of December 31, 2021 | $ | $ | $ | $ | $ | ||||||||||||
| Cash flows | — | ( | — | ( | |||||||||||||
| Non cash-flow: | |||||||||||||||||
| New leases | — | — | |||||||||||||||
| Foreign exchange adjustments | — | ( | ( | ( | |||||||||||||
| Other | — | ( | |||||||||||||||
| Liabilities as of December 31, 2022 | $ | $ | $ | $ | $ | ||||||||||||
| Cash flows | — | ( | — | — | ( | ||||||||||||
| Non cash-flow: | |||||||||||||||||
| New leases | — | ||||||||||||||||
| Foreign exchange adjustments | — | ||||||||||||||||
| Other | — | ( | |||||||||||||||
| Liabilities as of December 31, 2023 | $ | $ | $ | $ | $ | ||||||||||||
16.Restricted cash and other non-current financial assets
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
Restricted cash | ||||||||
Lease deposits and guarantees | ||||||||
Other non-current assets | ||||||||
Total | $ | $ | ||||||
Restricted cash is mainly related to deposits held against the Group's new office in Uppsala, Sweden.
17. Inventories
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
| Raw materials | $ | $ | ||||||
| Work in-progress | ||||||||
| Finished products | ||||||||
| Total inventories at the lower of cost and net realizable value | $ | $ | ||||||
The Group periodically analyses its inventory levels to identify inventory that may expire prior to expected usage or no longer meets quality specifications. When we have identified inventories to be in excess or obsolete, we write down the
F-34
value of those inventories to their net realizable value based upon judgment and estimates about future demand and market conditions. A provision for slow-moving and obsolete inventory is made within Cost of revenue. As of December 31, 2022 the provision amounted to $707 thousand. As per December 31, 2023 the provision amounted to $841 thousand.
18. Trade receivables
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
| Current | $ | $ | ||||||
| 1-30 days past due | $ | |||||||
| 31-60 days past due | $ | |||||||
| 61-90 days past due | $ | |||||||
| 91+ days past due | $ | |||||||
| Gross carrying amount | $ | $ | ||||||
| Allowance for expected credit losses | ( | ( | ||||||
| Net carrying amount | $ | $ | ||||||
Trade receivables, for the Group, are non-interest bearing and are generally on terms of 30 days to 90 days.
The Group maintains an allowance for ECL based on primarily historical data together with a forward-looking assessment but the Group have historically recognized almost non-existent credit losses. Of the total allowance of for expected credit losses as of December 31, 2023, $1,492 thousand is attributable to a single customer. This allocation reflects our evaluation of the credit risk associated with that customer's outstanding receivable balance.
The Group maintains an allowance for ECL based on primarily historical data together with a forward-looking assessment but the Group have historically recognized almost non-existent credit losses. Of the total allowance of for expected credit losses as of December 31, 2023, $
The credit loss recognized in the Company periods ended December 31, 2023 and December 31, 2022 was $1,909 and $236 thousand, respectively.
19. Other receivables
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
| Value added tax and other tax receivables | $ | $ | ||||||
| Other items | ||||||||
| Total | $ | $ | ||||||
20. Share capital and Other contributed capital
As of December 31, 2023, the total number of authorized shares was 400,000,000 of which 124,342,715 were issued and outstanding. During 2023, 5,010,253 shares were issued associated with the public offering launched in January and 234,344 shares were issued associated with the vesting of RSUs in the incentive award plan.
The Company’s Share capital and Other contributed capital at December 31, 2023 consisted of the following:
| Number of shares | Share Capital (amounts in thousands USD) | Other Contributed Capital (amounts in thousands USD) | |||||||||
| Common Shares | |||||||||||
| Total | |||||||||||
F-35
The Company’s Share capital and Other contributed capital at December 31, 2022 consisted of the following:
| Number of shares | Share Capital (amounts in thousands USD) | Other Contributed Capital (amounts in thousands USD) | |||||||||
| Common Shares | |||||||||||
| Total | |||||||||||
Initial public offering
On March 29, 2021, the Company completed an initial public offering of 13,235,294 ADSs, representing 13,235,294 common shares, at an initial public offering price of $20.00 per share. The net proceeds from the initial public offering were $249.3 million, after deducting the underwriting discounts, net of deferred taxes, and other initial public offering costs associated with the filing. The net proceeds of the initial public offering per the consolidated statement of cash flows of $245.2 million do not reflect the non-cash movement related to the tax-deductible portion of the underwriter fees. Total transaction costs accounted for as a deduction from equity, net of deferred taxes, amounts to $15.4 million.
Following the initial public offering on March 29, 2021 the Company had 119,007,062 common shares outstanding.
New share issue and public offering
(A)New share issue
On March 29, 2022, the Company issued 91,056 shares, associated with the vesting of Restricted stock units ("RSU") in the incentive award plan.
(B)Public offering
On January 18, 2023 the Company launched a public offering of 5,831,028 ADS each representing one common share of the Company (the “ADSs”), consisting of 4,250,000 ADSs offered by the Company and 1,581,028 ADSs offered by certain selling shareholders of the Company (the “Selling Shareholders”). In addition, the Company granted the underwriters a 30-day option to purchase up to 874,654 additional ADSs. The Company did not receive any proceeds from the sale of the ADSs by the Selling Shareholders. The offering closed on January 23, 2023, with respect to the initial 4,250,000 ADSs offered by the company and 1,581,028 ADSs/shares offered by the selling stockholders. The option granted to the underwriters closed February 13, 2023 with a total of 760,253 ADSs offered by the company pursuant to the time period. The net proceeds from the offering were $96.2 million, after deducting the underwriting discounts, net of deferred taxes, and other public offering costs associated with the filing. The net proceeds of the public offering per the condensed consolidated statement of cash flows of $95.1 million do not reflect the non-cash movement related to the tax-deductible portion of the underwriter fees and other public offering costs.
(C)New share issue
On March 22, 2023, the Company issued 234,344 shares, associated with the vesting of RSUs in the incentive award plan. Following the new share issue, the Company has 124,342,715 shares outstanding.
The following chart shows a reconciliation of the movements in equity from December 31, 2021 through December 31, 2022 and from December 31, 2022 through December 31, 2023:
F-36
| Shares Outstanding (number) | Share Capital (amounts in thousands USD) | Other Contributed Capital (amounts in thousands USD) | |||||||||
| Balance as of December 31, 2021 | $ | $ | |||||||||
| New Share Issuance | |||||||||||
| Share based remuneration program | — | ||||||||||
| Balance as of December 31, 2022 | $ | $ | |||||||||
| New Share Issuance | |||||||||||
| Share based remuneration program | |||||||||||
| Balance as of December 31, 2023 | $ | $ | |||||||||
21. Stock-based compensation
On April 17, 2023 at the Annual General Meeting, our shareholders resolved to adopt two long term incentive programs, LTI I 2023 and LTI II 2023 and simultaneously amending our Amended and Restated 2021 Incentive Award Plan (the “Plan”). The amendment to the plan increased the maximum shares of stock available for issuance by 980,000 shares. The 2021 Incentive Award Plan was initially adopted by the company on March 16, 2021, and approved by the shareholders of the Company on March 16, 2021, in connection with approval by the Company’s shareholders of LTI 2021 (the “Original Plan”). The Original Plan was amended and restated on April 7, 2022 at the Annual General Meeting when our shareholders resolved to adopt two long-term incentive programs, LTI I 2022 and LTI II 2022. The principal purpose of the Plan is to attract, retain and motivate selected employees, consultants and directors through the granting of share-based compensation awards and cash-based performance bonus awards. The Company has prior to 2023 filed two registration statements on Form S-8 covering 1,085,900 shares under the Original Plan and an additional 594,403 common shares under the Amended and Restated 2021 Incentive Award Plan. Together with the amendment approved On April 17, 2023 a total of 2,660,303 shares are available for issuance pursuant to a variety of stock-based compensation awards, including stock option and restricted stock unit awards; provided, however, that no more than 2,660,303 additional shares may be issued. Shares available under all plans will, subject to the terms and conditions of the Plan, be issued when the awards under the respective program vest, subject to continued service, over a four-year period from the grant date, and, in case of stock options, upon the option holder exercising the option. The expiration date on stock options awarded under the programs is five years from grant date.
Incentive stock options
In connection with the closing of the initial public offering, the Company granted options to purchase an aggregate of 620,675 common shares out of the Original Plan, of which 442,789 options were granted to certain executive officers and directors, in each case with an exercise price equal to 125 % of the initial public offering price of $20.00 . During the second quarter of 2022, 107,074 options that had been approved at the Annual General Meeting on April 7, 2022, were awarded to certain executive officers and directors, in each case with an exercise price of $17.39 which is equal to 100% of the share price at grant date. During the second quarter of 2023, 99,480 options that had been approved at the Annual General Meeting on April 17, 2023, were awarded to certain executive officers and directors, in each case with an exercise price of $22.79 which is equal to 100% of the share price at grant date.
Such options shall vest over four years , subject to the terms and conditions of the Plan. The expiration date on the options is five years from grant date.
The share-based compensation cost is calculated according to the following: The employee stock options were granted free of charge and are accounted for as equity-settled share-based payment transactions. Fair value per option at grant date multiplied by the number of outstanding share options multiplied by the number of days passed and divided by the total number of days in the vesting period. To calculate fair value per share option at the grant date, the principles of the Black-Scholes model have been used. The expense associated with these stock options amounted to $0.9 million for the twelve months ended December 31, 2023. The expense associated with these stock options amounted to $1.2 million for the twelve months ended December 31, 2022.
The following table lists the inputs to the Black-Scholes models used for stock options for the years ended December 31, 2023, and 2022.
F-37
| 2023 | 2022 | |||||||
| Expected volatility (%) | ||||||||
| Risk-free interest rate (%) | ||||||||
| Expected life of stock options (years) | ||||||||
| Share price at grant (US$) | ||||||||
A summary of stock option activity under the Company's Option Plans relating to awards to certain officers and directors as of December 31, 2023, and changes during the twelve months ended December 31, 2023 and December 31, 2022 , are as follows:
| Outstanding Stock Options | Weighted Average Exercise Price (USD) | |||||||
| Balance as of January 1, 2023 | $ | |||||||
| Granted | ||||||||
| Forfeited | ( | |||||||
| Balance as of December 31, 2023 | ||||||||
| Outstanding Stock Options | Weighted Average Exercise Price (USD) | |||||||
Balance as of January 1, 2022 | $ | |||||||
| Granted | ||||||||
| Forfeited | ||||||||
Balance as of December 31, 2022 | ||||||||
Restricted Stock Units
During 2022, 20,458 RSUs that had been approved at the Annual General Meeting on March 16, 2021 were awarded to employees currently employed by Olink under the 2021 Plan. During 2022, 607,866 RSUs that had been approved at the Annual General Meeting on April 7, 2022 were awarded to employees currently employed by Olink under the Plan. During 2023, 754,115 RSUs that had been approved at the Annual General Meeting on April 17, 2023 were awarded to employees currently employed by Olink under the Plan. 1,285,100 RSUs are outstanding as of December 31, 2023, of which 281,064 RSUs to our executive officers. The RSUs are measured based on the fair market value of the underlying ordinary shares on the date of grant. The RSUs will vest during a four-year period and new shares will be issued when the RSU’s vest.
The expense associated with these RSUs amounted to $12.6 million for the twelve months ended December 31, 2023. The expense associated with these RSUs amounted to $9.1 million for the twelve months ended December 31, 2022
The following is a summary of the RSU activity and related information as of December 31, 2023, and changes during the twelve months ended December 31, 2023 and December 31, 2022:
| Outstanding RSU's | Weighted Average Grant Date Fair Value (USD) | |||||||
| Balance as of January 1, 2023 | $ | |||||||
| Granted | ||||||||
| Vested | ( | |||||||
| Forfeited | ( | |||||||
| Balance as of December 31, 2023 | ||||||||
F-38
| Outstanding RSU's | Weighted Average Grant Date Fair Value (USD) | |||||||
Balance as of January 1, 2022 | $ | |||||||
| Granted | ||||||||
| Vested | ( | |||||||
| Forfeited | ( | |||||||
Balance as of December 31, 2022 | ||||||||
22. Other current liabilities
| Amounts in thousands USD | As of December 31, 2023 | As of December 31, 2022 | ||||||
| Other financial liabilities | ||||||||
| - Salaries and wages | $ | $ | ||||||
| - Royalties | ||||||||
| - Accrued expenses | ||||||||
| Other current liabilities | ||||||||
| Contract liabilities | ||||||||
| - Advances from customers | ||||||||
| Total | $ | $ | ||||||
Advance invoiced customers represent a contract liability. Beginning January 1, 2023, the Company had a liability balance of $1,694 thousand for advance invoiced customers. During fiscal year 2023, the Company recognized $1,500 thousand of the advances from invoiced customers as revenue. Beginning January 1, 2022, the Company had a liability balance of $5,447 thousand for advance invoiced customers. During fiscal year 2022, the Company recognized $5,342 thousand of the advances from customers as revenue.
23. Related-party transactions
Other than compensation arrangements with executive officer and directors, we have not entered into any material transactions with our executive officers, directors or holders, including their affiliates or other related parties during 2021, 2022 or 2023.
Compensation of key management personnel of the Group
| Amounts in thousands USD | |||||||||||
| For the year ended December 31, 2023 | For the year ended December 31, 2022 | For the year ended December 31, 2021 (1) | |||||||||
| Wages and salaries | $ | $ | $ | ||||||||
| Share-based compensation expense | |||||||||||
| Variable / bonus expense | |||||||||||
| Pension costs - defined contribution plans | |||||||||||
| $ | $ | $ | |||||||||
(1) For years ending December 31, 2021, we have restated to show separately variable / bonus expense, include share-based compensation expense, and exclude social charges expense.
F-39
Agreements with Our Executive Officers and Directors
Board members were paid for their services on the board of directors, board members collectively received remuneration of $408 thousand during the year ended December 31, 2021, $598 thousand during 2022 and $620 thousand during 2023.
Management Service Agreements
In 2021, the Summa MSA was terminated in connection with our initial public offering, upon which we paid Summa Equity AB a lump sum amount equal to approximately $2.4 million.
24. Earnings per share
Earnings per share for the Group is calculated by taking the net loss for the period, less the amount of the accumulated preferred dividend yield, divided by the weighted average of outstanding common shares during the period.
| for the year ended December 31, 2023 | for the year ended December 31, 2022 | for the year ended December 31, 2021 | |||||||||
| Net loss for the period | $ | ( | $ | ( | $ | ( | |||||
| Less accumulated preferred dividend yield | ( | ||||||||||
| Total | ( | ( | ( | ||||||||
| Weighted average number of shares (thousands) | |||||||||||
| Basic and diluted loss per share | $ | ( | $ | ( | $ | ( | |||||
As of December 31, 2021, the Group has the following potential common shares that can be potentially dilutive but are anti-dilutive for the periods presented and are therefore excluded from the weighted average number of common shares for the purpose of diluted profit/(loss) per share:
i.442,789 outstanding stock options related to the 2021 Incentive Award Plan (see note 21).
ii.335,449 restricted stock units related to the 2021 Incentive Award Plan (see note 21).
As of December 31, 2022, the Group has the following potential common shares that can be potentially dilutive but are anti-dilutive for the periods presented and are therefore excluded from the weighted average number of common shares for the purpose of diluted profit/(loss) per share:
i.549,863 outstanding stock options related to the 2021 Incentive Award Plan and the Amended and Restated 2021 Incentive Award Plan (see note 21).
ii.847,143 restricted stock units related to the 2021 Incentive Award Plan and the Amended and Restated 2021 Incentive Award Plan (see note 21).
As of December 31, 2023 the Group has the following potential common shares that can be potentially dilutive but are anti-dilutive for the periods presented and are therefore excluded from the weighted average number of common shares for the purpose of diluted profit/(loss) per share:
i.598,527 outstanding stock options related to the 2021 Incentive Award Plan and the Amended and Restated 2021 Incentive Award Plan (see note 21).
ii.1,285,100 restricted stock units related to the 2021 Incentive Award Plan and the Amended and Restated 2021 Incentive Award Plan (see note 21).
25. Subsequent events
F-40