UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
FOR ANNUAL AND TRANSITION REPORTS PURSUANT TO SECTIONS 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||
For the fiscal year ended |
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||
For the transition period from to |
(Commission file number)
(Exact name of registrant as specified in its charter)
State or other jurisdiction of incorporation or organization | (I.R.S. Employer Identification No.) |
|
|
|
|
(Address of principal executive offices) | (Zip Code) |
(
Registrant’s telephone number, including area code
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol(s) |
| Name of each exchange on which registered |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ◻
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ◻
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ |
☒ | Smaller reporting company | ||
|
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes
The aggregate market value of the voting common stock held by non-affiliates of the Registrant as of June 30, 2023 was approximately $
On March 1, 2024, approximately
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s proxy statement related to its 2024 Annual Stockholders’ Meeting to be filed subsequently are incorporated by reference into Part III of this Annual Report on Form 10-K. Except as expressly incorporated by reference, the registrant’s proxy statement shall not be deemed to be part of this report.
PROTALIX BIOTHERAPEUTICS, INC.
2023 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
PART I
Except where the context otherwise requires, the terms “we,” “us,” “our” or “the Company,” refer to the business of Protalix BioTherapeutics, Inc. and its consolidated subsidiaries, and “Protalix” or “Protalix Ltd.” refers to the business of Protalix Ltd., our wholly-owned subsidiary and sole operating unit.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
The statements set forth under the sections entitled “Business,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Risk Factors,” and other statements included elsewhere in this Annual Report on Form 10-K, particularly with respect to our plans and strategy for our business and related financing, include forward-looking statements within the meanings of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, including statements regarding expectations, beliefs, intentions or strategies for the future. When used in this report, the terms “anticipate,” “believe,” “estimate,” “expect,” “can,” “continue,” “could,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” and other words or phrases of similar import, as they relate to our company, our subsidiaries or our management, are intended to identify forward-looking statements. We intend that all forward-looking statements be subject to the safe-harbor provisions under the Private Securities Litigation Reform Act of 1995. These forward-looking statements are only predictions and reflect our views as of the date they are made with respect to future events and financial performance, and we undertake no obligation to update or revise, nor do we have a policy of updating or revising, any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events, except as may be required under applicable law. Forward-looking statements are subject to many risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements.
Examples of the risks and uncertainties include, but are not limited to, the following:
Given these uncertainties, you should not place undue reliance on these forward-looking statements. Companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced or late-stage clinical trials, even after obtaining promising earlier trial results or preliminary findings for such clinical trials. Even if favorable testing data is generated from clinical trials of a drug product, the FDA or foreign regulatory authorities may not accept or approve a marketing application filed by a pharmaceutical or biotechnology company for the drug product.
2
These and other risks and uncertainties are detailed under the “Risk Factors” section of this Annual Report and are described from time to time in the reports we file with the U.S. Securities and Exchange Commission, or the Commission.
Item 1.Business
We are a commercial stage biopharmaceutical company focused on the development, production and commercialization of recombinant therapeutic proteins produced via our proprietary ProCellEx® plant cell-based protein expression system. We are the first and only company to gain FDA approval of a protein produced through plant cell-based expression in suspension. Our unique expression system represents a new method for developing recombinant proteins in an industrial-scale manner.
To date, we have successfully developed two commercial products. On May 5, 2023, the European Commission, or EC, announced that it had approved the Marketing Authorization Application, or MAA, for Elfabrio and on May 9, 2023, the FDA announced that it had approved the Biologics License Application, or BLA, for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161, each for adult patients with a confirmed diagnosis of Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. Elfabrio was approved by the FDA with a boxed warning for hypersensitivity reactions/anaphylaxis, consistent with Enzyme Replacement Therapy (ERT) class labeling, and Warnings/Precautions providing guidance on the signs and symptoms of hypersensitivity and infusion-associated reactions seen in the clinical studies as well as treatments to manage such events should they occur. The Warnings/Precautions for membranoproliferative glomerulonephritis (MPGN) alert prescribers to the possibility of MPGN and provide guidance for appropriate patient management. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease. According to the EMA, overall, the benefit/risk balance of Elfabrio is positive in the claimed indication (Fabry disease). Elfabrio has also been approved for marketing in Great Britain, Switzerland and Israel.
The FDA publicly released the internal review documents for Elfabrio. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study (our phase I\II clinical trial of Fabry Disease, Study PB-102-F01/02) with confirmatory evidence provided by our phase III BALANCE clinical trial of PRX-102 for the treatment of Fabry disease, or the BALANCE study (also referred to as Study PB-102-F20). The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks. However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support a non-inferiority margin.
Our first product, Elelyso® (taliglucerase alfa, except in Brazil where it is marketed as BioManguinhos alfataliglicerase), an ERT for the treatment of patients with Gaucher disease was first approved by the FDA in May 2012 and is now approved for marketing in 23 markets including Brazil, Israel and others.
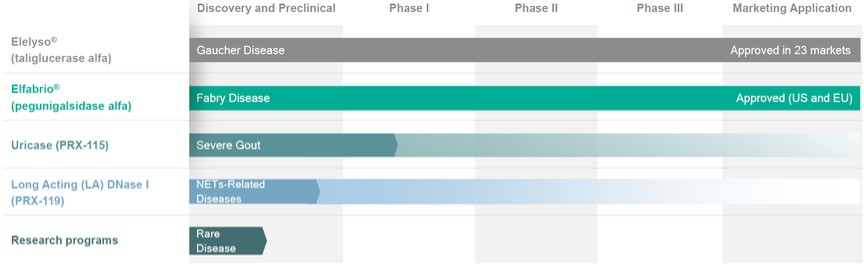
In addition, we are developing PEGylated uricase, or PRX-115, for the treatment of severe gout, Long Acting (LA) DNase I, or PRX-119, for the treatment of NETs-related diseases, and a number of other technologies and preclinical assets.
Our proprietary ProCellEx platform is being used to manufacture both of our approved and marketed products as well as PRX-115 and PRX-119.
Our strategy moving forward is to develop proprietary recombinant proteins designed to address high, unmet needs in the genetic and non-genetic rare disease space that are therapeutically superior to existing recombinant proteins currently marketed for the same indications. Consistent with this strategy, we have a number of product candidates in varying stages of the clinical development process.
3
Product Pipeline
ProCellEx: Our Proprietary Protein Expression System
ProCellEx is our proprietary platform used to produce and manufacture recombinant proteins through plant cell-based expression in suspension. ProCellEx consists of a comprehensive set of proprietary technologies and capabilities, including the use of advanced genetic engineering and plant cell culture technology, enabling us to produce complex, proprietary and biologically equivalent proteins for a variety of human diseases. Our protein expression system facilitates the creation and selection of high-expressing, genetically-stable cell lines capable of expressing recombinant proteins. We plan to execute on our strategy by developing tailored complex recombinant therapeutic proteins primarily produced through ProCellEx while genetically engineering and/or chemically modifying the proteins pre- and/or post-production. We intend such engineering and modifications to provide added clinical benefits by improving the biological characteristics (e.g., glycosylation, half-life, immunogenicity) of the therapeutic proteins.
Our ProCellEx technology allows for many unique advantages, including: biologic optimization; an ability to handle complex protein expressions; flexible manufacturing with improvements through efficiencies, enhancements and/or rapid horizontal scale-ups; a simplified production process; elimination of the risk of viral contaminations from mammalian components; and intellectual property advantages.
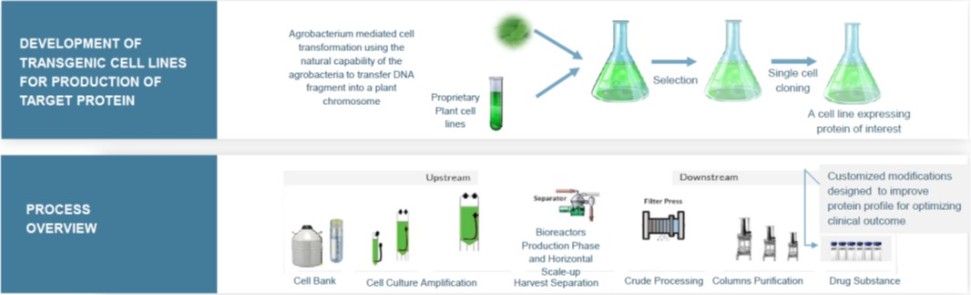
We developed ProCellEx based on our plant cell culture technology for the development, expression and manufacture of recombinant proteins which are the essential foundation of modern biotechnology. We develop new, recombinant therapeutic proteins by using the natural capability of agrobacterium to transfer a DNA fragment into the plant chromosome, allowing the genome of the plant cell to code for specific proteins of interest. The agrobacterium-mediated transformed cells are then able to produce specific proteins, which are extracted and purified and can be used as therapies to treat a variety of diseases. We are the first and only company to gain FDA approval of a protein produced through plant cell-based expression, and with the recent approval of Elfabrio, we now have two commercial proteins produced through our platform.
4
Our ProCellEx technology can be utilized to express complex therapeutic proteins belonging to different drug classes, such as enzymes, hormones, monoclonal antibodies, cytokines and vaccines. The entire protein expression process, from initial nucleotide cloning to large-scale production of the protein product, occurs under Current Good Manufacturing Practice-, or cGMP-, compliant, controlled processes. Our plant cell culture technology uses cells, such as carrot and tobacco (BY-2) cells, which undergo advanced genetic engineering and/or chemical modifications, and are grown on an industrial scale in a disposable, flexible bioreactor system. Our system does not involve mammalian or animal-derived components or transgenic field-grown or whole plants at any point in the production process.
Cell growth, from initiating scale-up steps from a cell-bank through large-scale production takes place in a clean-room environment in flexible, sterile, custom-designed polyethylene bioreactors, and does not require the use of large stainless-steel bioreactors commonly used in mammalian-based systems for recombinant protein production. The ProCellEx reactors are easy to use and maintain, allow for rapid horizontal scale-up and do not involve the risk of mammalian viral contamination. Our bioreactors are well-suited for plant cell growth using a simple, inexpensive, chemically defined growth medium. The reactors, which are custom-designed and optimized for plant cell cultures, require low initial capital investment and are rapidly scalable at a low cost.
Plant Cell Production Advantages

ProCellEx®: Protalix’s Differentiated Plant Cell Protein Expression Platform

5
Business Highlights
We have two commercial products, each of which is an ERT; Elelyso and Elfabrio. Our pipeline of product candidates includes PEGylated uricase for the treatment of severe gout, Long Acting (LA) DNase I for the treatment of NETs and other technologies and preclinical assets.
Elelyso® (taliglucerase alfa)
Elelyso for the treatment of Gaucher disease is currently approved and marketed in 23 countries including the United States, Australia, Canada, Israel, Brazil, Russia and Turkey. In June 2012, the EMA’s Committee for Medicinal Products for Human Use, or the CHMP, issued a positive opinion regarding the benefit of Elelyso but did not immediately grant marketing authorization because of the ten-year market exclusivity granted to Vpriv® (Takeda Shire) in August 2010 for the same condition, which was extended for an additional two years, and expired in August 2022. We have granted the marketing rights to Elelyso globally, excluding Brazil, to Pfizer through an exclusive licensing agreement. We maintain the distribution rights to Elelyso in Brazil, where it is currently marketed as BioManguinhos alfataliglicerase, through the Supply and Technology Transfer Agreement, or the Brazil Agreement, we entered into on June 18, 2013, with Fiocruz, an arm of the Brazilian MoH. In 2023, we generated $12.5 million from sales of Elelyso to Pfizer and $10.4 million from sales of BioManguinhos alfataliglicerase to the Brazilian MoH.
Elfabrio® (pegunigalsidase alfa orPRX-102)
Elfabrio for the treatment of adult patients with Fabry disease, is our proprietary, plant cell culture expressed enzyme, and a chemically modified stabilized version of, the recombinant α-Galactosidase-A protein, a lysosomal enzyme. Elfabrio has been approved by the FDA for marketing in the United States, the European Union, Great Britain, Switzerland and Israel. In 2023, we generated $17.5 million from sales of Elfabrio to Chiesi.
Protalix Ltd., our wholly-owned subsidiary, has entered into two exclusive licensing and supply agreements with Chiesi for Elfabrio, one global excluding the United States and the second for the United States.
Uricase (PRX-115)
PEGylated Uricase (PRX-115)
PRX-115 is our plant cell-expressed recombinant PEGylated Uricase (urate oxidase) – a chemically modified enzyme under development for the potential treatment of severe gout. Gout is the most common inflammatory arthritis, affecting an estimated 14.0 million adults in the United States, 2.0 million in France, 2.0 million in United Kingdom, 0.7 million in Italy, 1.5 million in Germany, 0.7 million in Spain and over 190.0 million in China. An estimated approximately 5% of the gout population is considered to have chronic refractory disease. Gout results from sustained elevation of serum urate levels (hyperuricaemia). Urate levels may increase due to diet, genetic predisposition and environmental factors leading to the deposition of monosodium urate crystals, tophi, in joints, tendons and other tissues, which triggers recurrent episodes of pronounced acute inflammation, known as gout flares. Gout leads to substantial morbidity, severe pain, reduced quality of life, decreased physical function, increased healthcare costs, and lost economic productivity. Furthermore, gout is strongly associated with a number of comorbidities, including hypertension, cardiovascular disease, renal impairment, diabetes, obesity, hyperlipidaemia and frequently in a combination known as the metabolic syndrome.
PRX-119
PRX-119 is our plant cell-expressed PEGylated recombinant human DNase I product candidate which we are designing to have an elongated half-life in the circulation for the potential treatment of NETs-related diseases. NETs, or Neutrophil extracellular traps, are web-like structures released by activated neutrophils that trap and kill a variety of microorganisms. NETs are composed of DNA, histones, antimicrobial and pro-inflammatory proteins. Excessive formation or ineffective clearance of NETs can cause different pathological effects. NETs formation has been observed in various autoimmune, inflammatory and fibrotic conditions, diverse forms of thrombosis, cancer and metastasis. According to scientific literature, animal studies have demonstrated that DNase I treatment reduce NETs toxicity. Our proprietary modified DNase I may potentially enable effective treatment for these conditions.
6
2023 and Recent Company Developments
Recent Developments
2023 Developments
7
Our Marketed Products
Elelyso for the Treatment of Gaucher Disease
Elelyso (taliglucerase alfa), our first commercial product, was approved by the FDA in 2012 for injection as an ERT for the long-term treatment of adult patients with a confirmed diagnosis of type 1 Gaucher disease. In August 2014, the FDA approved Elelyso for injection for pediatric patients. Elelyso is the first plant cell derived recombinant protein to be approved by the FDA for the treatment of Gaucher disease and is now approved in 23 markets including the United States, Brazil, Israel and others.
Gaucher disease, also known as glucocerebrosidase, or GCD, deficiency, is a rare genetic autosomal recessive disorder and one of the most common Lysosomal Storage Disorders, or LSDs, in the world. It is one of a group of disorders that affect specific enzymes that normally break down fatty substances for reuse in the cells. If the enzymes are missing or do not work properly, the substances can build up and become toxic. Gaucher disease occurs when a lipid called glucosylceramide accumulates in the cells of the bone marrow, lungs, spleen, liver, and sometimes the brain. Gaucher disease symptoms can include fatigue, anemia, easy bruising and bleeding, severe bone pain and easily broken bones, and distended stomach due to an enlarged spleen and thrombocytopenia. Epidemiology of Gaucher disease varies; Recent literature provides that prevalence of Gaucher disease ranges from 0.70 to 1.75 per 100,000 in the general population. In people of Ashkenazi Jewish heritage, estimates of occurrence vary from approximately 1 in 400 to 1 in 850 people.
The global market for Gaucher disease, that includes Sanofi’s Cerezyme®, Shire’s Vpriv® and Sanofi’s Cerdelga®, among others, was $1.6 billion in 2023, is forecasted to be approximately $1.6 billion in 2024 and is forecasted to grow at a CAGR of approximately 1% from 2023-2029.
The current standard of care for Gaucher disease is ERT, which is a medical treatment where recombinant enzymes are injected into patients to replace the lacking or dysfunctional enzyme. In Gaucher disease, recombinant GCD is infused to replace the mutated or deficient natural GCD enzyme. Elelyso is the only alternative ERT treatment of Gaucher disease to Sanofi Genzyme’s Cerezyme® and Takeda’s (Shire) Vpriv.
Elfabrio for the Treatment of Fabry Disease
Elfabrio, our second commercial product, was approved by the EC for marketing in the EU and by the FDA for marketing in the United States in May 2023 for adult patients with Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease. According to the EMA, overall, the benefit/risk balance of Elfabrio is positive in the claimed indication (Fabry disease). The FDA publicly released the internal review documents for Elfabrio. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study, our phase I\II clinical trial of Fabry disease, with confirmatory evidence provided by the BALANCE study. The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks. However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support the non-inferiority margin.
In August and September of 2023, Elfabrio was approved in Great Britain and Switzerland, respectively, and by the Israeli Ministry of Health in January 2024, for long-term enzyme replacement therapy in adult patients with a confirmed diagnosis of Fabry disease.
Fabry disease is a serious life-threatening rare genetic disorder. Fabry patients lack or have low levels of α-galactosidase-A resulting in the progressive accumulation of abnormal deposits of a fatty substance called globotriaosylceramide, or Gb3, in blood vessel walls throughout their body. The ultimate consequences of Gb3 deposition
8
range from episodes of pain and impaired peripheral sensation to end-organ failure, particularly of the kidneys, but also of the heart and the cerebrovascular system. Fabry disease occurs in one person per 40,000 to 60,000 males.
The standard of care for Fabry disease is ERT. Currently, the ERTs for Fabry disease are agalsidase alfa, agalsidase beta, and now Elfabrio. Through an ERT, the missing α-galactosidase-A is replaced with a recombinant form of the protein via intravenous, or IV, infusion once every two weeks. Fabry disease, if left untreated, will progress from a less severe condition to a more serious one. It can have a significant impact on quality of life due to presence of serious, chronic and debilitating complications, including cardiovascular and renal complications, and comorbid conditions such as pain can have a significant impact on the psychological well-being of Fabry patients and their social functioning. Fabry disease involves substantial reduction in life expectancy. Causes of death are most often cardiovascular disease and, to a lesser extent, cerebrovascular disease and renal disease. The life expectancy of Fabry patients is significantly shorter compared to the general population. Untreated male Fabry patients may experience shortened lifespans to approximately 50 years, and 70 years for untreated female patients with Fabry disease. This represents a 20- and 10-year reduction of their respective lifespans.
The global market for Fabry disease, that includes agalsidase beta, Sanofi’s Fabrazyme®, agalsidase alfa, Shire’s (acquired by Takeda Pharmaceutical Company Limited) Replagal® and Amicus Therapeutics’ Galafold®, among others, is forecasted to be approximately $2.0 billion in 2023 and is forecasted to grow at a CAGR of 6.8% from 2023-2029 reaching approximately $3.1 billion in annual sales at the end of the decade.
Regulatory Background
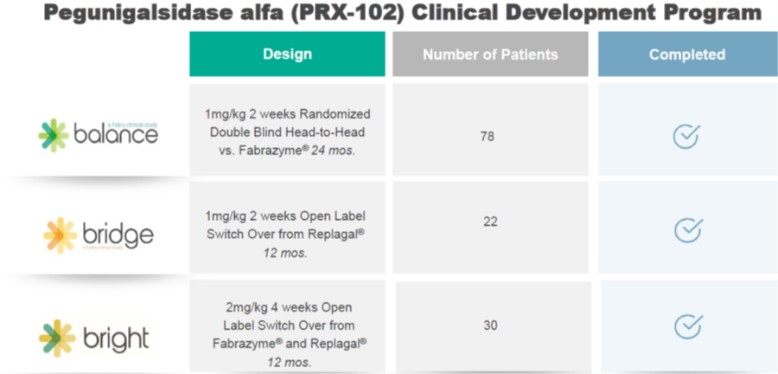
On November 9, 2022, we, together with Chiesi, resubmitted to the FDA a BLA for PRX-102, the name we assigned to Elfabrio internally prior to its approvals, for the potential treatment of adult patients with Fabry disease. The initial BLA for PRX-102 was submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, and the submission was subsequently accepted by the FDA and granted Priority Review designation. However, in April 2021, the FDA issued a complete response letter, or CRL, in response to the initial BLA. In preparation for the BLA resubmission, we and Chiesi participated in a Type A (End of Review) meeting with the FDA on September 9, 2021. As part of the meeting minutes provided by the FDA, which included the preliminary comments and meeting discussion, the FDA, in principle, agreed that the data package proposed to the FDA for a BLA resubmission had the potential to support a traditional approval of PRX-102 for the treatment of Fabry disease. The data package in the BLA resubmission, given the change in the regulatory landscape in the United States, included the final two-year analyses of our BALANCE study, which were completed in July 2022, and long-term data from our open-label extension study of PRX-102 in adult patients treated with a 2 mg/kg every four weeks dosage of PRX-102. The initial BLA included a comprehensive set of preclinical, clinical and manufacturing data compiled from our completed phase I/II clinical trial of PRX-102, including the related extension study, interim clinical data from our phase III BRIDGE clinical trial of PRX-102 for the treatment of Fabry disease, or the BRIDGE study, and safety data from our then on-going clinical studies of PRX-102 in adult patients receiving 1 mg/kg every two weeks.
The CRL did not report any concerns relating to the potential safety or efficacy of PRX-102 in the submitted data package. In the CRL, the FDA noted that an inspection of our manufacturing facility in Carmiel, Israel, including the FDA’s subsequent assessment of any related FDA findings, is required before the FDA can approve a new drug. Due to travel restrictions during the COVID-19 pandemic, the FDA was unable to conduct the required pre-approval inspection during the review cycle. In addition to the foregoing, the FDA noted that agalsidase beta had recently been converted to full approval, a change in regulatory circumstances which had to be addressed in the resubmitted BLA for PRX-102.
On February 7, 2022, the PRX-102 MAA was submitted to, and subsequently validated by, the EMA. The submission was made after the October 8, 2021 meeting we held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102. At the meeting, we and Chiesi discussed the scope of the anticipated MAA submission for the European Union, and the Rapporteur and Co-Rapporteur were generally supportive of the planned MAA submission. The MAA submission included a comprehensive set of preclinical, clinical and manufacturing data compiled from our completed and ongoing clinical studies evaluating PRX-102 as a potential alternative treatment for adult patients with Fabry disease. The submission was supported by the 12–month interim data analysis generated from our BALANCE study, which was released in June 2021. Data generated from the completed BRIDGE study, the phase I/II clinical trial in naive or untreated patients, and from the extension studies with 1 mg/kg every two weeks were also included in the
9
submission. In addition, the MAA included data from the completed 12–month switch–over phase III BRIGHT clinical trial of PRX-102 for the treatment of Fabry disease for adult patients with Fabry disease treated with the 2 mg/kg every four weeks dosage, or the BRIGHT study. As part of the EMA review process, we and Chiesi received the Day 120 list of questions in June 2022, and the full response package thereto was submitted to the EMA in September 2022 (following a 3-month clock-stop period). An essential portion of the response included the submission of the final analysis of the two-year BALANCE study (the final Clinical Study Report), and an interim analysis of the long-term, open-label extension study of PRX-102 in adult patients with Fabry disease treated with the 2 mg/kg every four weeks dosage.
On February 24, 2023, we, together with Chiesi, announced that the CHMP had adopted a positive opinion, recommending marketing authorization for PRX-102. The CHMP opinion was then referred for final action to the EC. As disclosed above, Elfabrio was subsequently approved by the EC for marketing in the EU and in the United States in May 2023 for adult patients with Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage.
Key Trials and Design
Our PRX-102 clinical development program was designed to show that PRX-102 has a potential clinical benefit in all adult Fabry patient populations when compared to the then marketed Fabry disease enzymes, agalsidase beta and agalsidase alfa. In preclinical studies, PRX-102 showed significantly longer half-life due to higher enzyme stability, enhanced activity in Fabry disease affected organs leading to reduction of the accumulated substrate and reduced immunogenicity, which together can potentially lead to improved efficacy through increased substrate clearance and significantly lower formation of anti-drug antibodies, or ADAs.
The phase III clinical program included three individual studies; the BALANCE study, the BRIDGE study the BRIGHT study, all of which have been completed. In the phase III clinical program overall, two potential dosing regimens for PRX-102 were analyzed; 1 mg/kg every two weeks, with the potential for improved efficacy and safety offering a potential alternative to existing enzyme replacement therapies, and 2 mg/kg every four weeks. The phase III program was preceded by the phase I/II clinical trial, a dose range finding study in ERT-naïve adult patients with Fabry disease, which was completed in 2016.
Phase III BALANCE Study
The BALANCE study (PB-102-F20, NCT02795676) was a pivotal 24-month, randomized, double blind, active control study of PRX-102 in adult Fabry patients with deteriorating renal function designed to evaluate the safety and efficacy of 1 mg/kg of PRX-102 administered every two weeks compared to agalsidase beta. The Clinical Study Report for the BALANCE study was completed in July 2022. The final analysis confirmed the positive top-line results (announced in April 2022) and favorable tolerability profile. A total of 78 patients who were previously treated with agalsidase beta for at least one year with an eGFR slope at screening worse than -2 mL/min/1.73 m2/year were enrolled in the study.
10
Patients were randomized on a 2:1 ratio for switching to PRX-102 or continuing on agalsidase beta. A total of 77 patients were treated; 52 with PRX-102 and 25 with agalsidase beta. Approximately 40% of the enrolled patients were female.
Forty-seven (90.4%) patients in the PRX-102 arm experienced at least one treatment-emergent adverse event (TEAE) compared to 24 (96.0%) in the agalsidase beta arm. The number of events adjusted to 100 years of exposure is 572.36 events for the PRX-102 arm and 816.85 events for the agalsidase beta arm.
TEAEs were reported for 21 (40.4%) patients in the PRX-102 arm compared to 11 (44.0%) in the agalsidase beta arm. The number of treatment-related events adjusted to 100 years of exposure is 42.85 events for the PRX-102 arm and 152.91 events for the agalsidase beta arm.
Usage of infusion pre-medication was tapered down during the study, if possible, for all patients. At baseline, 21 (40.4%) patients in the PRX-102 arm used infusion premedication compared to 16 (64.0%) in the agalsidase beta arm. At the end of the study, only three out of 47 (6.4%) patients in the PRX-102 arm used infusion premedication compared to three out of 24 (12.5%) in the agalsidase beta arm. Even with this reduction in use of premedication, there were fewer reported infusion-related reactions with PRX-102: 11 (21.2%) patients in the PRX-102 arm experienced a total of 13 events compared to six (24.0%) patients experiencing a total of 51 events in the agalsidase beta arm. The number of infusion-related reactions adjusted to 100 infusions is 0.5 for the PRX-102 arm and 3.9 for agalsidase beta arm.
Assessment of immunogenicity, that is, the existence and development of anti PRX-102 antibodies or anti-agalsidase beta antibodies, in the study indicated that for the PRX-102 arm, 18 (34.6%) patients were ADA positive at baseline, of which 17 (94.4%) had neutralizing antibody activity. For the agalsidase beta arm, eight (32.0%) patients were ADA positive at baseline, of which seven (87.5%) had neutralizing antibody activity. Only a small number of patients showed treatment-emergent ADA. At the end of the two-year study, 11 (23.4%) patients that received PRX-102 were ADA positive, of which seven (63.6%) had neutralizing antibody activity, while in the agalsidase beta arm six (26.1%) were ADA-positive and all six (100%) had neutralizing antibody activity. There was little change in the percentage of patients who were ADA positive, with a trend of reduction observed in the PRX-102 arm and stability in the agalsidase beta arm. The proportion of patients with neutralizing ADA decreased in the PRX-102 arm while it remained stable in the agalsidase beta arm.
Out of the 78 randomized patients, six patients discontinued the study: out of the five (9.4%) from the PRX-102 arm, one patient withdrew consent prior to the first infusion, two discontinued due to personal reasons, and two due to adverse events (one due to an unrelated adverse event and one due to a treatment related adverse event); one (4%) patient from the agalsidase beta arm discontinued for personal reasons. There were no deaths in this study.
Considering that in the trial patients in the PRX-102 arm were exposed for the first time to the novel enzyme, tolerability data appear favorable for PRX-102 and in-line with what was observed in the previous clinical studies of PRX-102.
Of the patients that completed the trial from both the PRX-102 and agalsidase beta treatment arms, 69 opted, with the advice of the treating physician, to receive PRX-102 1 mg/kg every two weeks in the long-term open-label extension study which is now sponsored by Chiesi.
The results of the direct, blinded comparison of PRX-102 to agalsidase beta, for the primary efficacy renal endpoints (i.e., eGFR change, eGFR slope) and for the main secondary endpoints (e.g., urine protein to creatinine ratio [UPCR] LVMI, MSSI, BPI) strongly suggest comparability in treatment effects between the two treatments.
At the same time a potentially favorable safety profile was identified based on lower rates of IRR, lower ADA positivity, and less premedication use in the PRX-102 arm compared to agalsidase beta. Overall, a positive benefit-risk balance was confirmed.
Phase III BRIDGE Study
The BRIDGE study (PB-102-F30, NCT03018730) was a 12-month open-label, single arm switch-over study evaluating the safety and efficacy of PRX-102, 1 mg/kg infused every two weeks, in up to 22 Fabry patients previously treated with
11
agalsidase alfa for at least two years and on a stable dose for at least six months. In the study, patients were screened and evaluated over three months while continuing agalsidase alfa treatment. The study was completed in December 2019.
Final results of the data generated in the BRIDGE study showed substantial improvement in renal function as measured by mean annualized eGFR slope in both male and female patients. Twenty of 22 patients completed the 12-month treatment duration. Eighteen of the patients who completed the study opted to roll over to a long-term extension study and continue to be treated with PRX-102. In the study, the mean annualized eGFR slope of the study participants improved from -5.90 mL/min/1.73m2/year while on agalsidase alfa to -1.19 mL/min/1.73m2/year on PRX-102 in all patients. Male patients improved from -6.36 mL/min/1.73m2/year to -1.73mL/min/1.73m2/year and female patients improved from -5.03 mL/min/1.73m2/year to -0.21 mL/min/1.73m2/year. Following the switch to PRX-102, there was a decrease in patients with progressing or fast progressing kidney disease which is consistent with the therapeutic goals for Fabry disease, as identified by Christoph Wanner, et. al., in 2019, and most patients achieved a stable status post-switch.
PRX-102 was well-tolerated in the BRIDGE study, with all adverse events being transient in nature without sequelae. Of the 22 patients enrolled in the BRIDGE study, the majority of TEAEs were mild or moderate in severity, with two patients (9.1%) withdrawing from the therapy due to hypersensitivity reaction that was resolved. The most common moderate TEAEs were nasopharyngitis, headache and dyspnea. An immunogenicity assessment indicated that four out of 20 patients (20%) developed persistent ADAs over the course of the study, of which two had neutralizing activity.
Of the patients that completed the trial, 18 opted, with the advice of the treating physician, to continue receiving PRX-102 1 mg/kg every two weeks in a long-term open-label extension study which is now sponsored by Chiesi.
Phase III BRIGHT Study
The BRIGHT study (PB-102-F50, NCT03180840) was a multicenter, multinational open-label, switch-over study designed to evaluate the safety, efficacy and pharmacokinetics of treatment with 2 mg/kg of PRX-102 administered every four weeks for 52 weeks (a total of 14 infusions). The study was completed in June 2020. The 2 mg/kg every four weeks dosage has not been approved by the EMA, FDA or any other jurisdiction.
Enrollment in the study included 30 adult patients (24 males and 6 females) with mean (SD) age of 40.5 (11.3) years, ranging from 19 to 58 years previously treated with a commercially available ERT (agalsidase beta or agalsidase alfa), for at least three years and on a stable dose administered every two weeks. To determine eligibility for participation in the study, candidates were screened to identify and select Fabry patients with clinically stable kidney disease. The most common Fabry disease symptoms at baseline were acroparesthesia, heat intolerance, angiokeratomas and hypohydrosis. Patients who matched the criteria were enrolled in the study and switched from their current treatment of IV infusions every two weeks to 2 mg/kg of PRX-102 every four weeks for 12 months. Patients participating in the study were evaluated, among other disease parameters, to determine if their kidney disease had not further deteriorated while being treated with the four-week dosing regimen as measured by eGFR and for lyso-Gb3 levels as a Fabry biomarker, as well as other parameters. In addition, participating patients were evaluated to assess the safety and tolerability of PRX-102.
The final results from the BRIGHT study indicate that 2 mg/kg of PRX-102 administered by intravenous infusion every four weeks was well tolerated, and Fabry disease assessed by eGFR slope and plasma lyso-Gb3 was stable throughout PRX-102 treatment in adult Fabry patients. None of the patients without ADAs at screening developed treatment-induced ADAs following the switch to PRX-102 treatment.
All 30 patients received at least one dose of PRX-102, and 29 patients completed the one-year study. Of these 29 patients, 28 received the intended regimen of 2 mg/kg every four weeks throughout the entire study, while one patient was switched to 1 mg/kg PRX-102 every two weeks per protocol at the 11th infusion. One patient withdrew from the study after the first infusion due to a traffic accident.
First infusions of PRX-102 were administered under controlled conditions at the investigation site. Based on the protocol-specified criteria, patients were able to receive their PRX-102 infusions at a home care setup once the applicable Investigator and Sponsor Medical Monitor agreed that it was safe to do so. Safety and efficacy exploratory endpoints were assessed throughout the 52-week study.
12
Overall, 33 of 183 total TEAEs reported in nine (30.0%) of the patients were considered treatment related; all were mild or moderate in severity and the majority were resolved at the end of the study. There were no serious or severe treatment-related TEAEs and no TEAEs led to death or study withdrawal. Of the treatment-related TEAEs, 27 were infusion-related reactions (IRRs) and the remainder were single events of diarrhea, erythema, fatigue, influenza-like illness, increases urine protein/creatinine ratio, and urine positive for white blood cells. The 27 IRRs were reported in five (16.7%) patients, all male. All IRRs occurred during the infusion or within two hours post-infusion; no events were recorded between two and 24 hours post-infusion.
The study suggests that Fabry patients who are currently receiving ERT every two weeks may be successfully transitioned to PRX-102 2 mg/kg every four weeks as an effective and tolerable alternative treatment option. Additional long term data is being collected as part of the ongoing long term extension study (PB-102-F51, NCT03614234) of the 2 mg/kg PRX-102 every four weeks dose.
Following a survey of participants using the Quality of Life EQ-5D-5L questionnaire, responses indicate that patient perception of their own health remained high and stable throughout the 52-week study duration, with overall health mean (SE) scores of 78.3 (3.1) and 82.1 (2.9) at baseline and Week 52, respectively, in a 0 to 100 scale. Using the short-form Brief Pain Inventory, or, questionnaire, approximately 75% of study participants had an improvement or no change in average pain severity at Week 52 (compared to baseline). The short-form BPI interference items also remained stable during the study. Pain-related results indicate that there was no increase and/or relapse in pain. No Fabry clinical events were reported during the study.
Of the patients that completed the trial, 29 opted, with the advice of the treating physician, to continue receiving PRX-102 2 mg/kg every four weeks in a long-term open-label extension study which is now sponsored by Chiesi. Two of such patients are being treated with 1 mg/kg every two weeks dosage.
Phase I/II Study
The phase I/II clinical trial of PRX-102 (NCT01678898) was a worldwide, multi-center, open-label, dose ranging study designed to evaluate the safety, tolerability, pharmacokinetics, immunogenicity and efficacy parameters of PRX-102 in adult patients with Fabry disease. It was completed in 2015.
We initiated the phase I\II study after PRX-102, in preclinical studies, showed significantly longer half-life due to higher enzyme stability, enhanced activity in Fabry disease affected organs leading to reduction of the accumulated substrate and reduced immunogenicity, which together can potentially lead to improved efficacy through increased substrate clearance and significantly lower formation of ADAs.
Sixteen adult, naïve Fabry patients (9 male and 7 female) completed the phase I/II study, each in one of three dosing groups, 0.2 mg/kg, 1 mg/kg and 2 mg/kg. Each patient received IV infusions of PRX-102 every two weeks for 12 weeks, with efficacy follow-up after six-month and twelve-month periods. The overall results demonstrate that PRX-102 reaches the affected tissue and reduces kidney Gb3 inclusions burden and lyso-Gb3 in the circulation. A high correlation was found between the two Fabry disease biomarkers, reduction of kidney Gb3 inclusions and the reduction of plasma lyso-Gb3 over six months of treatment.
Data was recorded at 24 months from 11 patients who completed 12 months of the long-term open-label extension trial that succeeded the phase I/II study. Patients who did not continue in the extension trial included female patients who became or planned to become pregnant and therefore were unable to continue in accordance with the study protocol and patients who relocated to a location where treatment was not available under the clinical study.
Results show that lyso-Gb3 levels decreased approximately 90% from baseline. Renal function remained stable with mean eGFR levels of 108.02 and 107.20 at baseline and 24 months, respectively, with a modest annual eGFR slope of -2.1. An improvement across all the gastrointestinal symptoms evaluated, including severity and frequency of abdominal pain and frequency of diarrhea, was noted. Cardiac parameters, including LVM, LVMI and EF, remained stable with no cardiac fibrosis development detected. In conclusion, an improvement of over 40% in disease severity was shown as measured by the Mainz Severity Score Index, or MSSI, a score compiling the different elements of the disease severity
13
including neurological, renal and cardiovascular parameters. In addition, an improvement was noted in each of the individual parameters of the MSSI.
The majority of adverse events were mild-to-moderate in severity, and transient in nature. During the first 12 months of treatment, only three of 16 patients (less than 19%) formed ADAs of which two of these patients (less than 13%) had neutralizing antibodies. Importantly, however, the ADAs turned negative for all three of these patients following 12 months of treatment. The ADA positivity effect had no observed impact on the safety, efficacy or continuous biomarker reduction of PRX-102.
Of the patients who completed the trial, 10 patients opted to continue receiving PRX-102 in an open-label, 60-month extension study under which all patients were switched to receive 1 mg/kg of the drug, the selected dose for our BALANCE and BRIDGE studies.
Extension Studies
Patients who completed the BALANCE, BRIDGE and BRIGHT studies, and the extension of the phase I/II study, were offered the opportunity to continue PRX-102 treatment in one of two long-term open-label extension studies. Overall, 126 subjects who participated in our PRX-102 clinical program initially opted, with the advice of the treating physician, to enroll in one of our long-term, open label, extension studies of PRX-102: 97 patients in the 1 mg/kg every two weeks extension study (PB-102-F60, NCT03566017) (10 subjects who completed an extension study from the phase I/II study, 18 subjects who completed the BRIDGE study; 69 subjects who completed the BALANCE study), and 29 subjects who completed the BRIGHT study in the 2 mg/kg every four weeks extension study (PB-102-F51, NCT03614234). Two of the subjects in the PB-102-F51 study are being treated with 1 mg/kg every two weeks. As of March 1, 2023, sponsorship of the two extension studies was transferred to Chiesi, and Chiesi is now administering the open-label extension studies.
Over time, and as Elfabrio is approved for marketing in different jurisdictions, participants switch-out of the open-label extension studies. Most of them have transferred to a commercial setting; others withdraw for other reasons.
Pediatric FLY Study
The pediatric FLY study, to be sponsored by Chiesi with our collaborative efforts, is currently in the start-up phase. The study is planned to be a multi-center, open-label trial to assess the safety, pharmacodynamics, efficacy and pharmacokinetics of Elfabrio in patients from two years to less than 18 years of age with confirmed Fabry disease. The design of the study is based on the Initial Pediatric Study Plan (iPSP) agreed to with the FDA and the paediatric investigation plan (PIP) for Elfabrio, which has been accepted, as amended, by the Paediatric Committee (PDCO) of the EMA.
Japanese RISE Study
Chiesi is currently enrolling patients in its Japanese RISE clinical trial of PRX-102 for the treatment of Fabry disease in Japan.
PEGylated Uricase (PRX-115)
PRX-115 is our recombinant PEGylated uricase (urate oxidase) – a chemically modified enzyme under development for the potential treatment of severe gout patients. The uricase enzyme converts uric acid to allantoin, which is easily eliminated through urine and does not exist naturally in humans. This recombinant enzyme, expressed via our ProCellEx system, is designed to lower uric acid levels and improve clinical manifestation of the disease while having low immunogenicity and increased half-life of the drug in the blood. Pre-clinical data demonstrates long half-life, reduced immunogenic risk and high specific activity which supports the potential of PRX-115 to be a safe and effective treatment for patients with gout. One-month multiple dosing toxicity studies in two species and 6-month multiple dosing toxicity study in one species were conducted to support single and multiple dose studies is humans.
A phase I First in Human (FIH) single ascending dose (SAD) double-blind, placebo-controlled study designed to evaluate the safety, pharmacokinetics, pharmacodynamics (reduction of uric acid) and immunogenicity of PRX-115 in patients with elevated uric acid levels (NCT05745727) was commenced in March 2023. The study is being conducted at
14
New Zealand Clinical Research (NZCR) under the New Zealand Medicines and Medical Devices Safety Authority (MedSafe) and the Health and Disability Ethics Committee (HDEC) guidelines. Fifty-six patients with no previous exposure to PEGylated uricase have been enrolled in the study. We anticipate announcing preliminary results in the second quarter of 2024.
Gout is the most common inflammatory arthritis, affecting an estimated 14.0 million adults in the United States, 2.0 million in France, 2.0 million in United Kingdom, 0.7 million in Italy, 1.5 million in Germany, 0.7 million in Spain and over 190.0 million in China. An estimated approximately 5% of the gout population is considered to have chronic refractory disease. The risk of gout increases with age, and it is thus more common in ageing populations. Gout results from sustained elevation of serum urate levels (hyperuricaemia). Urate levels may increase due to diet, genetic predisposition and environmental factors leading to the deposition of monosodium urate crystals and\or tophi in joints, tendons and other tissues, which triggers recurrent episodes of pronounced acute inflammation, known as gout flares. Gout leads to substantial morbidity, severe pain, reduced quality of life, decreased physical function, increased healthcare costs, and lost economic productivity. Furthermore, gout is strongly associated with a number of comorbidities, including hypertension, cardiovascular disease, renal impairment, diabetes, obesity, hyperlipidaemia and frequently in a combination known as the metabolic syndrome.
Severe gout is generally described as a state of gout in which there is a presence of monosodium urate crystals with any of the following: frequent recurrent gout flares, chronic gouty arthritis, subcutaneous tophi or disease elements of gout seen via imaging. Currently available urate-lowering therapies, can be effective in treating gout. However, low adherence, under dosing and disease progression that cause high patient burden require new, effective and safe therapies to treat these severe, underserved gout patients.
To date, two variants of recombinant uricases are approved for marketing: (i) Krystexxa® (pegloticase) for treatment of chronic gout refractory to conventional therapy (gout patients who have contraindication/failure of other lowering uric acid treatments) and (ii) Elitek®, indicated for the treatment of tumor lysis syndrome but not gout. Both have a black box warning for anaphylaxis and other major side-effects. In particular, 89% of patients treated with Krystexxa developed an immunogenic response associated with a failure to maintain normalization of serum uric acid levels over a 6-month therapy cycle. The FDA label of krystexxa was amended in 2022 to include co-treatment of metrotrexate to prolong efficacy and increases tolerability in patients with refractory gout. Krystexxa is no longer marketed in the European Union. The EC withdrew the marketing authorization for Krystexxa in 2016 at the request of the marketing authorization holder which notified the EC of its decision not to market the product in the European Union for commercial reasons. We believe that new effective, safe therapies are needed to treat severe gout, chronic refractory and uncontrolled gout, regardless of treatment history.
PRX-119
PRX-119 is our plant cell-expressed PEGylated recombinant human DNase I product candidate being designed to elongate half-life in the circulation for NETs-related diseases. NETs, Neutrophil extracellular traps, are web-like structures, released by activated neutrophils that trap and kill a variety of microorganisms. NETs are composed of DNA, histones, antimicrobial and pro-inflammatory proteins. Excessive formation or ineffective clearance of NETs can cause different pathological effects. NETs formation has been observed in various autoimmune, inflammatory and fibrotic conditions, diverse forms of thrombosis, cancer and metastasis. According to scientific literature, animal studies have demonstrated that DNase treatment reduces NETs toxicity. Our proprietary modified DNase I design for long and customized systemically circulating in the bloodstream, may potentially enable effective treatment of these conditions.
The administration of PRX-119 resulted in a decrease in circulating of DNA levels and significantly enhanced the survival of mice in both a CLP-induced sepsis model and an ARDS model.
Intellectual Property
We have a robust patent portfolio, which is a key element of our overall strategy. We work to continually enhance, strengthen, and protect our intellectual property and now hold a broad portfolio of approximately 90 patents globally, including in Europe, the United States, Israel and additional countries worldwide. Our patents are designed to protect our
15
proprietary technology, proprietary products and product candidates, and their methods of use. Additionally, we have approximately 50 pending patent applications.
During the year ended December 31, 2023, we received the following:
| ● | New patents in each of New Zealand and the Russian Federation for the patent family named “Therapeutic Regimen For The Treatment of Fabry Using Stabilized Alpha-galactosidase, adding to the single previously granted patent in such family. |
| ● | A new patent in Brazil for the patent family named “DNase I Polypeptides, Polynucleotides Encoding Same, Methods of Producing DNase I and uses thereof in Therapy, adding to the two previously granted patents in such family. |
| ● | A new patent in Brazil for the patent family named “Nucleic Acid Construct for Expression of Alpha-Galactosidase in Plants and Plant Cells,” adding to the nine previously granted patents in such family. |
| ● | New patents in each of the USA, New Zealand, Israel and South Africa for the patent family named “Modified DNase and uses thereof,” adding to the three previously granted patents in such family. |
Our competitive position and future success depend, in part, on our ability, and that of our licensees, to obtain and leverage the intellectual property rights covering our product candidates, know-how, methods, processes and other technologies, to protect our trade secrets, to prevent others from using our intellectual property and to operate without infringing on the intellectual property rights of third parties. We seek to protect our competitive position by filing United States, European Union, Israeli and other foreign patent applications covering our technology, including both new technology and improvements to existing technology. Our patent strategy includes obtaining patents on methods of production, compositions of matter and methods of use. We also rely on know-how, continuing technological innovation, licensing and partnership opportunities to develop and maintain our competitive position.
Our outstanding 7.50% Senior Secured Convertible Notes due 2024, or the 2024 Notes, are guaranteed by our subsidiaries and secured by perfected liens on all of our material assets, primarily consisting of our intellectual property assets, including a stock pledge of our foreign subsidiaries in favor of the holders of outstanding 2024 Notes.
As of December 31, 2023, our patent portfolio consisted of several patent families (consisting of patents and/or patent applications) covering our technology, protein expression methodologies and system and product candidates, as follows:
Production of High Mannose Proteins in Plant Culture/PCT/ Il2004 000181 | N/A | Japan, Israel, Canada, Russian Federation, Mexico, India, Australia, South Africa, Republic of Korea, Singapore, Europe, Hong Kong, Ukraine, China, USA, Brazil | 2024(1) |
Plant Cell/Tissue Culturing Device, System and Method/PCT/Il2005/ 000228 | N/A | Israel | 2025 |
Large Scale Disposable Bioreactor/PCT/Il2008/000614 | N/A | Australia, Canada, China, Europe, Hong Kong, India, Israel, Republic of Korea, Russian Federation, Singapore, South Africa, USA, Brazil | 2028(2) |
Stabilized Alpha-galactosidase and uses thereof/PCT/Il2011/ 000209 | Brazil | Canada, South Africa, Russian Federation, Singapore, Israel, India, New Zealand, | 2031(3) |
16
China, Japan, USA, Europe, | |||
Nucleic Acid Construct for Expression of Alpha-galactosidase in Plants and Plant Cells/PCT/ Il2011/000719 | N/A | India, China, Republic of Korea, Japan, Israel, Europe, Hong Kong, USA, Brazil | 2024(2) |
Therapeutic Regimen For The Treatment of Fabry Using Stabilized Alpha-galactosidase/ PCT/Il2018/050018 | USA, Europe, Brazil, Japan, Canada, Australia, Chile, Israel, Republic of Korea, China, Mexico, Hong Kong | South Africa, New Zealand, Russian Federation | 2038 |
Dry Powder Formulations of DNase 1/PCT/Il2013/050094 | N/A | Israel, USA | 2033 |
DNase I Polypeptides, Polynucleotides Encoding Same, Methods of Producing DNase I and uses thereof in Therapy/PCT/ Il2013/050097 | N/A | Europe, Israel, Brazil | 2033 |
Inhalable Liquid Formulations of DNase I/PCT/Il2013/050096 | N/A | Israel, USA | 2033 |
Modified DNase and uses thereof/ PCT/Il2016/050003 | Europe, Canada, China, | USA, Australia, Mexico, Israel, | 2036 |
Use of Plant Cells Expressing a TNF Alpha Polypeptide Inhibitor in Therapy/PCT/IL2014/050231 | N/A | Israel | 2034 |
Removal of Constructs from Transformed Cells/PCT/IL2019/ 051266 | USA, Israel, Japan, New Zealand, Australia | N/A | N/A |
Long-Acting DNase/PCT/IL2021/051207 | Canada, Israel, USA, Japan, Europe, Hong Kong, Republic of Korea, China | N/A | N/A |
Dicer-Like Knock-Out Plant Cells/ PCT/IL2021/051194 | Israel, USA, Japan, Europe, Hong Kong, Republic of Korea, China | N/A | N/A |
Modified Uricase and Uses Thereof/PCT/IL2021/051305 | Japan, Canada, Brazil, USA, Israel, Mexico, Europe, Republic of Korea, China | N/A | N/A |
Methods of treating diseases associated with elevated uric acid | N/A | N/A | N/A |
| (1) | Patent granted in Australia expires in 2029. |
| (2) | Patent granted in the United States expires in 2032. |
| (3) | PTE/SPC applications were submitted for some of the patents. |
We are aware of U.S. patents, and corresponding international counterparts of such patents, owned by third parties that contain claims covering methods of producing glucocerebrosidase. We do not believe that, if any claim of infringement were to be asserted against us based upon such patents, taliglucerase alfa would be found to infringe any valid claim under such patents. However, there can be no assurance that a court would find in our favor or that, if we choose or are required to seek a license to any one or more of such patents, a license would be available to us on acceptable terms or at all.
In April 2005, Protalix Ltd. entered into a license agreement with Icon Genetics AG, or Icon, pursuant to which we received an exclusive worldwide license to develop, test, use and commercialize Icon’s technology to express certain proteins in our ProCellEx protein expression system. We are also entitled to a non-exclusive worldwide license to make and have made other proteins expressed by using Icon’s technology. As consideration for the license, we are obligated to make royalty payments equal to varying low, single-digit percentages of net sales of products by us, our affiliates, or any
17
sublicensees under the agreement. In addition, we are obligated to make milestone payments equal to $350,000, in the aggregate, for each product developed under the license, upon the achievement of certain milestones.
Our license agreement with Icon remains in effect until the earlier of the expiration of the last patent under the agreement or, if all of the patents under the agreement expire, 20 years after the first commercial sale of any product under the agreement. Icon may terminate the agreement upon written notice to us that we are in material breach of our obligations under the agreement and we are unable to remedy such material breach within 30 days after we receive such notice. Further, Icon may terminate the agreement in connection with certain events relating to a wind up or bankruptcy, if we make a general assignment for the benefit of our creditors, or if we cease to conduct operations for a certain period. Icon may also terminate the exclusivity granted to us by written notice if we fail to reach certain milestones within a designated period of time. Notwithstanding the termination date of the agreement, our obligation to pay royalties to Icon under the agreement may expire prior to the termination of the agreement, subject to certain conditions.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly evolving technology and significant competition. Competition from numerous existing companies and others entering the fields in which we operate is intense and expected to increase. Most of these companies have substantially greater research and development, manufacturing, marketing, financial, technological personnel and managerial resources than we do. In addition, many specialized biotechnology companies have formed collaborations with large, established companies to support research, development and commercialization of products that may be competitive with our current and future product candidates and technologies. Acquisitions of competing companies by large pharmaceutical or biotechnology companies could further enhance such competitors’ financial, marketing and other resources. Academic institutions, governmental agencies and other public and private research organizations are also conducting research activities and seeking patent protection and may commercialize competitive products or technologies on their own through collaborations with pharmaceutical and biotechnology companies.
With respect to Gaucher disease, we face competition primarily from two ERTs, Sanofi Genzyme’s Cerezyme and Takeda’s (Shire) Vpriv. In addition, Actelion markets a small molecule drug for the treatment of mild to moderate Type 1 Gaucher disease (Zavesca or miglustat), an oral treatment approved by the FDA only for patients for whom ERT is not a therapeutic option. In addition, Sanofi Genzyme markets a small molecule oral drug, Cerdelga®, approved for Gaucher patients with certain CYP2D6 metabolizer status. We are aware of other treatments and gene therapies in early clinical development and later stage clinical development for the treatment of Gaucher disease.
With respect to Fabry disease, we face competition primarily from Sanofi Genzyme (Fabrazyme), Takeda (Replagal) and Amicus (Galafold®). In addition, we are aware of other late clinical stage, early clinical stage and experimental drugs that are being developed by other companies for the treatment of Fabry disease.
With respect to severe gout, we face competition from Horizon Therapeutics Public Limited Company (Krsytexxa), which is indicated for treatment of chronic gout in adult patients refractory to conventional therapy. In addition, we are aware of other clinical stage, early clinical stage and experimental refractory or chronic gout treatments. For example, we are aware of a product candidate for chronic refractory gout that recently completed a phase III clinical trial and has met the primary endpoints of the trial, and of another product candidate that is the subject of a phase II clinical trial for hyperuricemia in gout patients with advanced CKD.
We also face potential competition to our ProCellEx system from companies that are developing other platforms for the expression of recombinant therapeutic pharmaceuticals. We are aware of companies that are developing alternative technologies to develop and produce therapeutic proteins in anticipation of the expiration of certain patent claims covering marketed proteins. A number of companies have developed or are developing alternative expression technologies. Examples include Crucell N.V.’s (acquired by Johnson & Johnson) expression system based on human-cell technology, Dyadic International Inc.’s expression system based on a fungus, Pfenex Inc.’s (acquired by Ligand Pharmaceuticals Incorporated) bacteria-based expression system, and others. Companies developing alternate plant-based technologies include iBio, Inc., Medicago, Inc., and eleva. Unlike ProCellEx, these alternate technologies are not cell-based. These companies base their product development on transgenic plants or whole plants.
18
Agreements and Partnerships
Elelyso – Pfizer
We have licensed to Pfizer the global rights to Elelyso in all markets, excluding Brazil, pursuant to an Amended and Restated Exclusive License and Supply Agreement, or the Amended Pfizer Agreement, which we entered into with Pfizer in October 2015 to amend and restate our initial Exclusive License and Supply Agreement with Pfizer, or the Pfizer Agreement. Pursuant to the Amended Pfizer Agreement, Pfizer retains 100% of revenue and reimburses 100% of direct costs. For the first 10-year period after the execution of the Amended Pfizer Agreement, we have agreed to sell drug substance to Pfizer for the production of Elelyso, subject to certain terms and conditions, and Pfizer maintains the right to extend the supply period for up to two additional 30-month periods, subject to certain terms and conditions. In a subsequent amendment, we agreed that after the completion of the first 10-year supply period, the supply term would automatically extend for a five-year period. Any failure to comply with our supply commitments may subject us to substantial financial penalties. The Amended Pfizer Agreement includes customary provisions regarding cooperation for regulatory matters, patent enforcement, termination, indemnification and insurance requirements. We retain distribution rights to taliglucerase alfa in Brazil.
Elelyso – Fundação Oswaldo Cruz (Fiocruz)
Elelyso, marketed as BioManguinhos alfataliglicerase in Brazil, is commercialized in Brazil through the Brazil Agreement with Fiocruz, which became effective in January 2014. Gaucher patients in Brazil are entitled to receive ERT paid for by the Brazilian MoH. The Brazilian MoH clinical treatment guidelines (PCDT) state that BioManguinhos alfataliglicerase is the therapy of choice for newly diagnosed patients. BioManguinhos alfataliglicerase is currently estimated to be used by approximately 25% of Gaucher patients in Brazil.
The Brazil Agreement provides for a staged technology transfer which is intended to transfer to Fiocruz the capacity and skills required for the Brazilian government to construct its own manufacturing facility, at its sole expense, and to produce a sustainable, high-quality, and cost-effective supply of BioManguinhos alfataliglicerase. Fiocruz has not satisfied certain purchase commitments under the Brazil Agreement. Accordingly, we and Fiocruz discuss on a continuous basis, potential steps to maximize sales of BioManguinhos alfataliglicerase sales to the Brazilian MoH.
Elfabrio (pegunigalsidase alfa\PRX-102) – Chiesi Farmaceutici
We have entered into two exclusive global licensing and supply agreements for PRX-102 for the treatment of Fabry disease with Chiesi; the Exclusive License and Supply Agreement dated as of October 17, 2017, made by and between Protalix Ltd. and Chiesi, or the Chiesi Ex-US Agreement, and the Exclusive License and Supply Agreement dated as of July 23, 2018, made by and between Protalix Ltd. and Chiesi, or the Chiesi US Agreement. The Chiesi Ex-US Agreement and the Chiesi US Agreement are referred to herein collectively as the Chiesi Agreements. Under the agreements, Protalix Ltd. has received $50.0 million in upfront payments and development cost reimbursements of $45 million, and is entitled to more than $1.0 billion in potential milestone payments tiered payments for drug product purchased from Protalix Ltd. equal to 15% - 35% (ex-US) and 15% - 40% (US), depending on the amount of annual net sales in the applicable territories.
Under the Chiesi Ex-US Agreement, we granted to Chiesi an exclusive license for all markets outside of the United States to commercialize PRX-102. Chiesi made an upfront payment to Protalix Ltd. of $25.0 million in connection with the execution of the agreement, and Protalix Ltd. was entitled to additional payments of up to $25.0 million in development costs in the aggregate, capped at $10.0 million per year. Protalix Ltd. is also eligible to receive additional payments of up to a maximum of $320.0 million, in the aggregate, in regulatory and commercial milestone payments. Protalix Ltd. agreed to manufacture all of the PRX-102 needed for all purposes under the agreement, subject to certain exceptions, and Chiesi will purchase PRX-102 from Protalix Ltd., subject to certain terms and conditions. Chiesi is required to make tiered payments of 15% to 35% of its net sales, depending on the amount of annual sales, as consideration for the supply of PRX-102.
The exclusive license to develop and commercialize PRX-102 in the United States were granted to Chiesi under the Chiesi US Agreement. Protalix Ltd. received an upfront, non-refundable, non-creditable payment of $25.0 million from
19
Chiesi and was entitled to additional payments of up to a maximum of $20.0 million to cover development costs for PRX-102, capped at $7.5 million per year. Protalix Ltd. is also eligible to receive additional payments of up to a maximum of $760.0 million, in the aggregate, in regulatory and commercial milestone payments. Chiesi agreed to make tiered payments of 15% to 40% of its net sales under the Chiesi US Agreement to Protalix Ltd., depending on the amount of annual sales, subject to certain terms and conditions, as consideration for product supply.
On May 13, 2021, we signed a binding term sheet with Chiesi amending the Chiesi Agreements in order to provide our company with near-term capital. Chiesi agreed to make a $10.0 million payment to us before the end of the second quarter of 2021 in exchange for a $25.0 million reduction in a longer term regulatory milestone payment provided in the Chiesi EX-US Agreement. All other regulatory and commercial milestone payments remain unchanged. We received the payment in June 2021. We also agreed to negotiate certain manufacturing related matters.
On August 29, 2022, we entered into the F/F Agreement and the Letter Agreement. Under the F/F Agreement, we agreed to supply Chiesi with drug substance for PRX-102 and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide us with commercial fill/finish services for PRX-102, including to support the anticipated global launch of PRX-102. The F/F Agreement will expire December 31, 2025, unless terminated earlier in accordance with its terms and may be extended by mutual agreement in writing for an additional period of seven years. The Letter Agreement changed the obligations of both us and Chiesi under the Chiesi Agreements with respect to, among other things, the evaluation, selection and establishment of an initial alternate source of commercial fill/finish services for PRX-102. In addition, the Letter Agreement amended certain provisions of the Chiesi Agreements to reflect the appointment of Chiesi as a supplier to our company of commercial fill/finish services and the potential establishment of an initial alternate source of commercial fill/finish services.
Manufacturing
We use our current manufacturing facility in Carmiel, Israel, which has approximately 14,700 sq/ft of clean rooms built according to industry standards, to manufacture drug substance for Elfabrio and Elelyso for commercial purposes and, with respect to Elfabrio, in connection with clinical trials. We maintain an approximately 3,400 sq/ft pilot plant for protein development and to manufacture supplies for clinical trials (phase I and phase II). Elelyso, Elfabrio and our drug product candidates must be manufactured in a clean environment and in compliance with cGMPs set by the FDA and other relevant foreign regulatory authorities. We intend to use our manufacturing space to produce all of the drug substance needed in connection with the clinical trials for our other product candidates.
In 2017, the FDA approved the supplemental New Drug Application (sNDA) we submitted to allow us to convert our manufacturing facility from a single dedicated product facility to a multi-product facility. This conversion allows us to realize potentially significant operational savings. Our facility’s current capacity can serve all of our current and expected commercial and clinical needs.
Our manufacturing facilities are subject to inspections by various regulatory authorities from time to time. We have undergone successful inspections by the FDA, the Irish Medicines Board (under the EMA’s centralized marketing authorization procedure), the Brazilian National Health Surveillance Agency (ANVISA), the Israeli Ministry of Health, the Turkish Ministry of Health, the Australian Therapeutic Goods Administration (TGA) and Health Canada.
Our current facility in Israel was granted “Approved Enterprise” status, and we have elected to participate in the alternative benefits program. Our facility is located in a top priority location, or “Zone A,” location, and, therefore, our income from the Approved Enterprise will be tax exempt in Israel for a 10-year period, commencing with the year in which we first generate taxable income from the relevant Approved Enterprise and after we use our net operating loss carryforwards, or NOLs. We expect to be entitled to similar tax benefits for a number of years thereafter. To remain eligible for these tax benefits, we must continue to meet certain conditions, and if we increase our activities outside of Israel, for example, by future acquisitions, such increased activities generally may not be eligible for inclusion in Israeli tax benefit programs. In addition, our technology is subject to certain restrictions with respect to the transfer of technology and manufacturing rights.
20
Raw Materials and Suppliers
We believe that the raw materials that we require throughout the manufacturing process of Elfabrio and Elelyso, and our current and potential drug product candidates, are widely available from numerous suppliers and are generally considered to be generic industrial biological supplies. We rely on a single, approved supplier for certain materials relating to the current expression of our proprietary biotherapeutic proteins through ProCellEx. We have identified additional suppliers for most of the materials required for the production of our product candidates.
Development and regulatory approval of our pharmaceutical products are dependent upon our ability to procure active ingredients (DS) and certain packaging materials from sources approved by the FDA and other regulatory authorities. The FDA and other regulatory approval processes require manufacturers to specify their proposed suppliers of active ingredients (which is not relevant to our company), intermediate material and certain packaging materials in their applications. From time to time, we intend to continue to identify alternative approved suppliers to ensure the uninterrupted supply of necessary raw materials.
Government Regulations
U.S. Drug Development Process
The FDA regulates drugs under the U.S. Federal Food, Drug, and Cosmetic Act, or the FFDCA, and its implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. Biologics are subject to regulation by the FDA under the FFDCA, the Public Health Service Act, and related regulations and other federal, state and local laws and regulations. Biological products include a wide variety of products including vaccines, blood and blood components, gene therapies, tissue and proteins. Unlike most prescription products made through chemical processes, biological products generally are made from human and/or animal materials. To be lawfully marketed in interstate commerce, a biologic product must be the subject of a BLA issued by the FDA on the basis of a demonstration that the product is safe, pure and potent, and that the facility in which the product is manufactured meets standards to assure that the product continues to be safe, pure and potent. The FDA has developed, and is continuously updating, the requirements with respect to cell and gene therapy products and has issued documents concerning the regulation of cellular and tissue-based products. Manufacturers of cell and tissue-based products must comply with the FDA’s current good tissue practices, or cGTP, which are FDA regulations that govern the methods used in, and the facilities and controls used for, the manufacture of such products. The primary intent of the cGTP requirements is to ensure that cell and tissue-based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease.
The process of obtaining regulatory approvals and ensuring compliance with appropriate federal, state and local statutes and regulations in the United States, and foreign statutes and regulations, requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, product detention, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. The process required by the FDA before a biological product or drug may be marketed in the United States generally involves the following:
| ● | Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices or other regulations; |
| ● | Submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| ● | Performance of adequate and well-controlled clinical trials according to Good Clinical Practices, or GCP, to establish the safety and efficacy of the proposed biological product or drug for its intended use; |
21
| ● | Submission to the FDA of a BLA for a new biological product or a new drug application, or NDA, for a new drug; |
| ● | Satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with cGMP to assure that the facilities, methods and controls are adequate to preserve the drug’s or biologic’s identity, strength, quality and purity; and |
| ● | FDA review and approval of the BLA or NDA. |
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all subjects participating in the clinical trial provide their informed consent regarding the trial. Further, an institutional review board, or IRB, must review and approve the plan for any clinical trial before it commences at any institution. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject, or his or her legal representative, and must monitor the clinical trial until completed. Once an IND is in effect, each new clinical protocol and any amendments to the protocol must be submitted to the FDA for review, and to the IRBs for approval.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase I. The product is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients having the specific disease. |
| ● | Phase II. Phase II clinical trials involve investigations in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and the optimal dosage and schedule. |
| ● | Phase III. Phase III clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for regulatory approval and product labeling. |
Post-approval studies, also called Phase IV trials, may be conducted after initial marketing approvals. These studies are used to obtain additional experience from the treatment of patients in the intended therapeutic indication and may be required by the FDA as part of the approval process.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected side effects. Phase I, Phase II and Phase III testing may not be completed successfully within any specified period, if at all. The FDA or the trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the study subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product candidate has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the applicable product candidate does not undergo unacceptable deterioration over its shelf life.
22
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the product candidate, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA or BLA, requesting approval to market the product. The submission of an NDA or BLA is subject to the payment of substantial user fees which may be waived under certain limited circumstances.
The testing and approval processes require substantial time and effort, and may not result in an approval on a timely basis, if at all. The FDA may refuse to approve a BLA or NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Generally, it takes one to three years to obtain approval. If questions arise during the FDA review process, approval may take a significantly longer period of time.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase IV testing which involves clinical trials designed to further assess a drug’s or biologic’s safety and effectiveness after BLA or NDA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Orphan Drug Designation
Under the Orphan Drug Act of 1983, the FDA may grant orphan drug designation to drugs and biological products intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States or that affects more than 200,000 persons in the United States but that sales in the United States are not expected to recover the costs of developing and marketing a treatment drug. Orphan product designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Among the benefits of orphan drug designation are possible funding and tax savings to support clinical trials, other financial incentives and a waiver of the marketing application user fee.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same treatment for the same indication for seven years, except in limited circumstances, such as (i) the drug’s orphan designation is revoked; (ii) its marketing approval is withdrawn; (iii) the orphan exclusivity holder consents to the approval of another applicant’s product; (iv) the orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of clinical superiority to the product with orphan exclusivity by a competitor product. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan drug status in the European Union has similar but not identical benefits in the European Union.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA marketing approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between (a) the effective date of an IND and the submission date of a BLA or an NDA plus (b) the time between the submission date of a BLA or an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the extension must be requested prior to expiration of the patent. The U.S. Patent and Trademark Office, or USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. We anticipate that we will apply for restorations of the patent term for certain of patents covering our product candidates.
23
Fast Track Designation
The FDA has a fast track program that is designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need, the purpose being to make important new drugs available to patients earlier. A drug candidate that receives Fast Track designation from the FDA is eligible for some or all of the following: more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of appropriate data needed to support drug approval; more frequent written communication from the FDA about such things as the design of the proposed clinical trials; eligibility for the FDA’s Accelerated Approval and Priority Review, if relevant criteria are met; and eligibility for Rolling Review, which allows a drug company to submit completed sections of its BLA or NDA for review by the FDA, rather than waiting until every section of the BLA or NDA is completed before the entire application can be reviewed. BLA or NDA review usually does not begin until the drug company has submitted the entire application to the FDA. We used the Rolling Review option for our taliglucerase alfa NDA, which we completed in April 2010.
Accelerated Approval
Section 901 of the U.S. Food and Drug Administration Safety Innovations Act amends the FFDCA to allow the FDA to base Accelerated Approval for drugs for serious conditions that fill an unmet medical need on whether the drug has an effect on a surrogate or an intermediate clinical endpoint. A surrogate endpoint used for Accelerated Approval is a marker; that is, a laboratory measurement, radiographic image, physical sign or other measure, that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. An intermediate clinical endpoint is a measure of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a drug, such as an effect on irreversible morbidity and mortality. The FDA bases its decision on whether to accept the proposed surrogate or intermediate clinical endpoint on the scientific support for that endpoint. Studies that demonstrate a drug’s effect on a surrogate or intermediate clinical endpoint must be “adequate and well controlled” as required by the FFDCA.
The Accelerated Approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a drug, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. Under subpart H of the Accelerated Approval pathway, the FDA may grant marketing approval for a new drug product on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely, based on epidemiologic, therapeutic, pathophysiologic, or other evidence, to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. The Accelerated Approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the drug’s clinical benefit. As a result, a drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase IV or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, would allow the FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
Post-Approval Requirements
Any drugs for which we receive FDA approval are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse effects with the product, reporting of changes in distributed products which would require field alert reports (FARs), drugs and biological product deviation reports (BPDRs) providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. In September 2007, the Food and Drug Administration Amendments Act of 2007 was enacted, giving the FDA enhanced post-marketing authority, including the authority to require post marketing studies and clinical trials (PMRs and PMCs), labeling changes based on new safety information, and compliance with risk evaluations and mitigation strategies (REMS), approved by the FDA. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Drugs and biologics may be promoted only for the approved indications and in accordance with the provisions of the approved label. Further, manufacturers of drugs and biologics must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, changes to the manufacturing process generally
24
require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Drug and biologic manufacturers and other entities involved in the manufacturing and distribution of approved drugs and biologics are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP, GTP applicable to biologics, and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the drug. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards, and test each product batch or lot prior to its release.
The FDA may withdraw a product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Discovery of previously unknown problems with a product subsequent to its approval may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, warning letters, holds on clinical trials, product recalls or seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions or civil or criminal penalties.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, the FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our development efforts. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
Foreign Regulation
We are subject to regulations and product registration requirements in many foreign countries in which we may sell our products, including in the areas of product standards, packaging requirements, labeling requirements, import and export restrictions and tariff regulations, duties and tax requirements. The time required to obtain clearance required by foreign countries may be longer or shorter than that required for FDA clearance, and requirements for licensing a product in a foreign country may differ significantly from FDA requirements.
Pharmaceutical products may not be imported into, or manufactured or marketed in, the State of Israel absent drug registration. The three basic criteria for the registration of pharmaceuticals in Israel is quality, safety and efficacy of the pharmaceutical product and the Israeli Ministry of Health requires pharmaceutical companies to conform to international developments and standards. Regulatory requirements are constantly changing in accordance with scientific advances as well as social and ethical values.
The relevant legislation of the European Union requires that medicinal products, including generic versions of previously approved products, and new strengths, dosage forms and formulations, of previously approved products, shall have a marketing authorization before they are placed on the market in the European Union. Authorizations are granted after the assessment of quality, safety and efficacy by the respective health authorities. In order to obtain an authorization, an application must be made to the competent authority of the member state concerned or in a centralized procedure to the EMA. Besides various formal requirements, the application must contain the results of pharmaceutical (physico-chemical, biological or microbiological) tests, of preclinical (toxicological and pharmacological) tests as well as of clinical trials. All of these tests must have been conducted in accordance with relevant EU regulations and must allow the reviewer to evaluate the quality, safety and efficacy of the medicinal product. Orphan drug designation in the European Union is granted to medicinal products intended for the diagnosis, prevention and treatment of life-threatening diseases and very serious conditions that affect not more than five in 10,000 people in the European Union. Orphan drug designation is generally given to medicinal products that treat conditions for which no current therapy exists or are expected to bring a significant benefit to patients over existing therapies.
25
Third Party Payor Coverage and Reimbursement
Coverage and reimbursement status of any approved therapy carries uncertainty and risk. In both the United States and foreign markets, our ability to commercialize our product and product candidates successfully, and to attract commercialization partners, depends in significant part on the availability of adequate financial coverage and reimbursement from third party payors, including, in the United States, governmental payors such as Medicare, Medicaid and the Veterans Affairs Health programs, and private health insurers. Medicare is a federally funded program managed by the Centers for Medicare and Medicaid Services, or CMS, through local fiscal intermediaries and carriers that administer coverage and reimbursement for certain healthcare items and services furnished to the elderly and disabled. Medicaid is an insurance program for certain categories of patients whose income and assets fall below state defined levels and who are otherwise uninsured that is both federally and state funded and managed by each state. The federal government sets general guidelines for Medicaid and each state creates specific regulations that govern its individual program. Each payor has its own process and standards for determining whether it will cover and reimburse a procedure or particular product. Private payors often rely on the lead of the governmental payors in rendering coverage and reimbursement determinations. Therefore, achieving favorable CMS coverage and reimbursement is usually a significant gating issue for successful introduction of a new product. The competitive position of some of our products will depend, in part, upon the extent of coverage and adequate reimbursement for such products and for the procedures in which such products are used. Prices at which we or our customers seek reimbursement for our product candidates can be subject to challenge, reduction or denial by the government and other payors.
Possible legislation at the federal and state levels in the United States focused on cost containment and price transparency may impact our ability to sell our product and product candidates for maximum profitably. It appears likely that the pressure on pharmaceutical pricing will continue, especially under the Medicare program, which may also increase our regulatory burdens and operating costs. Moreover, additional changes could be made to governmental healthcare programs that could significantly impact the success of our product and product candidates.
Some third party payors also require pre-approval of coverage for new or innovative devices, biologics or drug therapies before they will reimburse healthcare providers that use such therapies. While we cannot predict whether any proposed cost-containment measures will be adopted or otherwise implemented in the future, the announcement or adoption of these proposals could have a material adverse effect on our ability to obtain adequate prices for our product candidates and operate profitably.
Different pricing and reimbursement schemes exist in other countries. In the European Union, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs and biologics, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney General offices within the Department of Justice, and state and local governments. These regulations include:
| ● | the federal healthcare program anti-kickback law, which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
26
| ● | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other government reimbursement programs that are false or fraudulent; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
| ● | the federal transparency requirements under the Health Care Reform Law requires manufacturers of drugs, devices, biologics, and medical supplies to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests; |
| ● | the FFDCA, which among other things, strictly regulates drug and biologic product marketing, prohibits manufacturers from marketing drug products for off-label use and regulates the distribution of drug samples; and |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts. |
Compliance with Environmental, Health and Safety Laws
In addition to FDA regulations, we are also subject to evolving federal, state and local environmental, health and safety laws and regulations. In the past, compliance with environmental, health and safety laws and regulations has not had a material effect on our capital expenditures. Compliance with environmental, health and safety laws and regulations in the future may require additional capital expenditures.
Israeli Government Programs
The following is a brief summary of the current principal Israeli tax laws applicable to us and Protalix Ltd., and of the Israeli Government programs from which Protalix Ltd. benefits. Some parts of this discussion are based on new tax legislation that has not been subject to judicial or administrative interpretation. Therefore, the views expressed in the discussion may not be accepted by the tax authorities in question. This summary is based on laws and regulations in effect as of the date hereof, and should not be construed as legal or professional tax advice and does not cover all possible tax considerations.
General Corporate Tax Structure in Israel
The income of Protalix Ltd., other than income from “Approved Enterprises,” is taxed in Israel at regular rates. Pursuant to the Economic Efficiency Law (Legislative Amendments for Implementing the Economic Policy for the 2017 and 2018 Budget Year), 2016, the corporate tax rate in 2018 and thereafter is 23%. Capital gains on the sale of assets are subject to capital gains tax according to the corporate tax rate in effect in the year which the assets are sold.
Law for the Encouragement of Capital Investments, 1959
The Law for the Encouragement of Capital Investments, 1959, as amended, or the Investment Law, provides certain incentives for capital investments in a production facility (or other eligible assets). Generally, an investment program that is implemented in accordance with the provisions of the Investment Law, referred to as an “Approved Enterprise,” is entitled to benefits. These benefits may include cash grants from the Israeli government and tax benefits, based upon, among other things, the location within Israel of the facility in which the investment is made and specific elections made by the grantee. In order to qualify for these incentives, an Approved Enterprise is required to comply with the requirements of the Investment Law, and Letter of approval received by Protalix Ltd.
27
Protalix Ltd. will continue to enjoy the tax benefits under the pre-revision provisions of the Investment Law. If any new benefits are granted to Protalix Ltd. in the future, Protalix Ltd. will be subject to the provisions of the amended Investment Law with respect to these new benefits. Therefore, the following discussion is a summary of the Investment Law prior to its amendment as well as the relevant changes contained in the new legislation.
Under the Investment Law prior to its amendment, a company that wished to receive benefits had to receive approval from the Authority for the Investment and Development of the Industry and Economy, or the Investment Center. Each certificate of approval for an Approved Enterprise relates to a specific investment program in the Approved Enterprise, delineated both by the financial scope of the investment and by the physical characteristics of the facility or the asset, e.g., the equipment to be purchased and utilized pursuant to the program.
An Approved Enterprise may elect to forego any entitlement to the grants otherwise available under the Investment Law and, instead, participate in an alternative benefits program under which the undistributed income (after deductions and offsets) from the Approved Enterprise is exempt from corporate tax for a defined period of time. Under the alternative package of benefits, a company’s undistributed income derived from an Approved Enterprise will be exempt from corporate tax for a period of between two and 10 years from the first year of taxable income, depending upon the geographic location within Israel of the Approved Enterprise. Upon expiration of the exemption period, the Approved Enterprise is eligible for the reduced tax rates otherwise applicable under the Investment Law for any remainder of the otherwise applicable benefits period (up to an aggregate benefits period of either seven or 10 years, depending on the location of the company or its definition as a foreign investors’ company). If a company has more than one Approved Enterprise program or if only a portion of its capital investments are approved, its effective tax rate is the result of a weighted combination of the applicable rates. The tax benefits from any certificate of approval relate only to taxable profits attributable to the specific Approved Enterprise and are contingent upon meeting the criteria set out in the certificate of approval. Income derived from activity that is not integral to the activity of the Approved Enterprises (including capital gain) does not enjoy these tax benefits.
A company that has an Approved Enterprise program is eligible for further tax benefits, as an alternative to exemption, if it qualifies as a foreign investors’ company. A foreign investors’ company eligible for benefits is essentially a company in which more than 25% of the share capital (in terms of shares, rights to profit, voting and appointment of directors) is owned (measured by both share capital and combined share and loan capital) by non-Israeli residents. A company that qualifies as a foreign investors’ company and has an Approved Enterprise program is eligible for tax benefits for a 10-year benefit period and may enjoy a reduced corporate tax rate of 10% to 23%, depending on the amount of the company’s shares held by non-Israeli shareholders.
If a company that has an Approved Enterprise program is a wholly-owned subsidiary of another company, the percentage of foreign investments is determined based on the percentage of foreign investment in the parent company. The tax rates and related levels of foreign investments with respect to a foreign investor’s company that has an Approved Enterprise program are set forth in the following table:
Percent of Foreign Ownership |
| Rate of Reduced Tax |
Over 25% but less than 49% |
| 23% |
49% or more but less than 74% |
| 20% |
74% or more but less than 90% |
| 15% |
90% or more |
| 10% |
Our original facility in Israel has been granted “Approved Enterprise” status, and it has elected to participate in the alternative benefits program. Under the terms of its Approved Enterprise program, the facility is located in a Zone A area and, therefore, the undistributed income from that Approved Enterprise will be tax exempt in Israel for a period of 10 years, commencing with the year in which taxable income is first generated from the relevant Approved Enterprise. The current benefits program may not continue to be available and Protalix Ltd. may not continue to qualify for its benefits.
A company that has elected to participate in the alternative benefits program and that subsequently pays a dividend out of the income derived from the portion of its facilities that have been granted Approved Enterprise status during the tax exemption period will be subject to corporate tax in respect of the amount of dividend distributed at the rate that would
28
have been applicable had the company not elected the alternative benefits program (generally 10% to 23%, depending on the extent to which non-Israeli shareholders hold such company’s shares). If the dividend is distributed within 12 years after the end of the benefits period (or, in the case of a foreign investor’s company, without time limitation), the dividend recipient is taxed at the reduced withholding tax rate of 20% applicable to dividends from approved enterprises, or at the lower rate under an applicable tax treaty. After this period, the withholding tax rate is 25% to 30%, or at the lower rate under an applicable tax treaty. In the case of a company with a foreign investment level (as defined by the Investment Law) of 25% or more, the 12-year limitation on reduced withholding tax on dividends does not apply. The company must withhold this tax at its source, regardless of whether the dividend is converted into foreign currency.
The Investment Law also provides that an Approved Enterprise is entitled to accelerated depreciation on its property and equipment that are included in an approved investment program. This benefit is an incentive granted by the Israeli government regardless of whether the alternative benefits program is elected.
The benefits available to an Approved Enterprise are conditioned upon terms stipulated in the Investment Law and its regulations and the criteria set forth in the applicable certificate of approval. If Protalix Ltd. does not fulfill these conditions in whole or in part, the benefits can be canceled and Protalix Ltd. may be required to refund the benefits received, linked to the Israeli consumer price index with interest. We believe that Protalix Ltd. currently operates in compliance with all applicable conditions and criteria.
Amendment No. 60 to the Investment Law introduced a tax benefits regime referred to as “Benefitted Enterprises.” Under the Investment Law, the approval of the Investment Center is required only for Benefitted Enterprises that receive cash grants. Benefitted Enterprises that do not receive benefits in the form of governmental cash grants, but only tax benefits, are no longer required to obtain this approval. Instead, these Benefitted Enterprises are required to make certain investments as specified in the Investment Law.
The amended Investment Law specifies certain conditions for a Benefitted Enterprise to be entitled to benefits. These conditions include, inter alia, the following:
| ● | the Benefitted Enterprise’s revenues from any single country or a separate customs territory may not exceed 75% of the Benefitted Enterprise’s total revenues; or |
| ● | at least 25% of the Benefitted Enterprise’s revenues during the benefits period must be derived from sales into a single country or a separate customs territory with a population of at least 14 million people (starting from January 1, 2012, 1.4% must be added for each year). |
There can be no assurance that Protalix Ltd. will comply with the above conditions in the future or that Protalix Ltd. will be entitled to any additional benefits under the Investment Law. In addition, it is possible that Protalix Ltd. may not be able to operate in a manner that maximizes utilization of the potential benefits available under the Investment Law.
In the future there may be changes in the law, subject to the preservation of benefits, which may affect the benefits available to companies under the Investment Law. The termination or substantial reduction of any of the benefits available under the Investment Law could impact our tax expenses.
Amendment of the Law for the Encouragement of Capital Investments, 1959
In recent years, several amendments have been made to the Investments Law which have enabled new alternative benefit tracks, subject to certain conditions. The Investments Law was amended as part of the Economic Policy Law for the years 2011-2012 (amendment 68 to the Encouragement of Capital Investments Law), which was passed by the Israeli Knesset on December 29, 2010. The amendment sets alternative benefit tracks to those currently in effect under the provisions of the Investments Law. On December 29, 2016, Amendment 73 to the Investments Law, or the Investments Law Amendment, was published. This amendment sets new benefit tracks, inter alia, “Preferred Technological Enterprise” and “Special Preferred Technological Enterprise.” To date, we have elected not to have the Investments Law Amendment apply to our company.
29
Encouragement of Industrial Research, Development and Technology Innovation Law, 1984
To date, Protalix Ltd. has received grants from the Office of the Chief Scientist of the Israeli Department of Labor, or the OCS, under the Israeli Law for the Encouragement of Industrial Research, Development and Technology Innovation, 1984, and related regulations, or the Research Law. On January 1, 2016, the Israeli government established the National Authority for Technological Innovation, or NATI, which replaced many of the functions of the OCS. For purposes of clarity, references to NATI will include the OCS. NATI grants may be made available, from time to time, to finance a portion of Protalix Ltd.’s research and development expenditures in Israel. As of December 31, 2023, NATI approved grants in respect of Protalix Ltd.’s continuing operations totaling approximately $53.2 million (before interest, as described below), measured from inception. Protalix Ltd. is required to repay up to 100% of grants actually received (plus interest at the LIBOR rate applied to the grants received on or after January 1, 1999) to NATI through payments of royalties at a rate of 3% to 6% of the revenues generated from NATI-funded project, depending on the period in which revenues were generated. As of December 31, 2023, Protalix Ltd. either paid or accrued royalties payable of $17.7 million, and Protalix Ltd.’s contingent liability to NATI with respect to grants received was approximately $35.5 million.
Under the Research Law, recipients of grants from NATI are prohibited from manufacturing products developed using these grants outside of the State of Israel without special approvals, although the Research Law does enable companies to seek prior approval for conducting manufacturing activities outside of Israel without being subject to increased royalties. If Protalix Ltd. receives approval to manufacture the products developed with government grants outside of Israel, it will be required to pay an increased total amount of royalties (possibly up to 150% of the grant amounts plus interest), depending on the manufacturing volume that is performed outside of Israel, as well as at a possibly increased royalty rate.
Additionally, under the Research Law, Protalix Ltd. is prohibited from transferring NATI-financed technologies and related intellectual property rights outside of the State of Israel, except under limited circumstances and only with the approval of NATI Council or the Research Committee. Protalix Ltd. may not receive the required approvals for any proposed transfer and, if received, Protalix Ltd. may be required to pay NATI a portion of the consideration that it receives upon any sale of such technology by a non-Israeli entity. The scope of the support received, the royalties that Protalix Ltd. has already paid to NATI, the amount of time that has elapsed between the date on which the know-how was transferred and the date on which NATI grants were received and the sale price and the form of transaction will be taken into account in order to calculate the amount of the payment to NATI. The payments to NATI may result in up to 600% of the grant amounts plus interest. Approval of the transfer of technology to residents of the State of Israel is required, and may be granted in specific circumstances only if the recipient abides by the provisions of applicable laws, including the restrictions on the transfer of know-how and the obligation to pay royalties. No assurance can be made that approval to any such transfer, if requested, will be granted.
Under the Research Law and the regulations promulgated thereunder, the NATI Council may allow the transfer outside of Israel of know-how derived from an approved program and the related manufacturing rights in limited circumstances which are currently as follows:
| ● | in the event of a sale of know-how itself to a non-affiliated third party, provided that upon such sale the owner of the know-how pays to NATI an amount, in cash, as set forth in the Research Law (and the regulations promulgated thereunder). In addition, the amendment provides that if the purchaser of the know-how gives the selling Israeli company the right to exploit the know-how by way of an exclusive, irrevocable and unlimited license, the research committee may approve such transfer in special cases without requiring a cash payment. |
| ● | in the event of a sale of a company which is the owner of know-how, pursuant to which the company ceases to be an Israeli company, provided that upon such sale, the owner of the know-how makes a cash payment to NATI as set forth in the Research Law (and the regulations promulgated thereunder). |
| ● | in the event of an exchange of know-how such that in exchange for the transfer of know-how outside of Israel, the recipient of the know-how transfers other know-how to the company in Israel in a manner in |
30
| which NATI is convinced that the Israeli economy realizes a greater, overall benefit from the exchange of know-how. |
The Research Committee may, in special cases, approve the transfer of manufacture or of manufacturing rights of a product developed within the framework of the approved program or which results therefrom, outside of Israel.
The State of Israel does not own intellectual property rights in technology developed with NATI funding and there is no restriction on the export of products manufactured using technology developed with NATI funding. The technology is, however, subject to transfer of technology and manufacturing rights restrictions as described above. For a description of such restrictions, please see “Risk Factors—Risks Relating to Our Operations in Israel.” NATI approval is not required for the export of any products resulting from the research or development or for the licensing of any technology in the ordinary course of business.
Law for the Encouragement of Industry (Taxes), 1969
We believe that Protalix Ltd. currently qualifies as an “Industrial Company” within the meaning of the Law for the Encouragement of Industry (Taxes), 1969, or the Industry Encouragement Law. The Industry Encouragement Law defines “Industrial Company” as a company resident in Israel and incorporated in Israel, that derives 90% or more of its income in any tax year (other than specified kinds of passive income such as capital gains, interest and dividends) from an “Industrial Enterprise” operating in Israel (including Judea & Samaria territories and the Gaza strip), that it owns. An “Industrial Enterprise” is defined as an enterprise whose major activity in a given tax year is industrial production.
The following corporate tax benefits, among others, are available to Industrial Companies:
| ● | amortization of the cost of purchased know-how and patents over an eight-year period for tax purposes; |
| ● | accelerated depreciation rates on equipment and buildings; |
| ● | under specified conditions, an election to file consolidated tax returns with other related Israeli Industrial Companies; and |
| ● | expenses related to a public offering are deductible in equal amounts over three years. |
Eligibility for the benefits under the Industry Encouragement Law is not subject to receipt of prior approval from any governmental authority. It is possible that Protalix Ltd. may fail to qualify or may not continue to qualify as an “Industrial Company” or that the benefits described above will not be available in the future.
Tax Benefits for Research and Development
Under specified conditions, Israeli tax laws allow a tax deduction by a company for research and development expenditures, including capital expenditures, for the year in which such expenditures are incurred. These expenditures must relate to scientific research and development projects and must be approved by NATI. Furthermore, the research and development projects must be for the promotion of the company and carried out by or on behalf of the company seeking such tax deduction. However, the amount of such deductible expenditures is reduced by the sum of any funds received through government grants for the finance of such scientific research and development projects. Research and development expenses which were not approved shall be deductible over a period of three years.
Employees
As of December 31, 2023, we had 208 employees of whom 208 are full time employees and 16 have a Ph.D. or an M.D. in their respective scientific fields. We believe that our relations with our employees are good. We believe that our success will greatly depend on our ability to identify, attract and retain capable employees. The Israeli Ministry of Labor and Welfare is authorized to make certain industry-wide collective bargaining agreements, or Expansion Orders, that apply to types of industries or employees including ours. These agreements affect matters such as cost of living adjustments to salaries, length of working hours and week, recuperation, travel expenses, and pension rights. Otherwise, our employees are not represented by a labor union or represented under a collective bargaining agreement. See “Risk
31
Factors—We depend upon key employees and consultants in a competitive market for skilled personnel. If we are unable to attract and retain key personnel, it could adversely affect our ability to develop and market our products.”
Company Background
We were originally incorporated in the State of Florida in April 1992, and reincorporated in the State of Delaware in March 2016. Protalix Ltd., our wholly-owned subsidiary and sole operating unit, is an Israeli company and was incorporated in Israel in 1993.
ProCellEx® is our registered trademark. Each of the other trademarks, trade names or service marks appearing in this Annual Report on Form 10-K belongs to its respective holder.
Available Information
Our main corporate website address is http://www.protalix.com. We make available on our website, free of charge, our Commission filings, including our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any amendments to these reports, as soon as reasonably practicable after we electronically file these documents with, or furnish them to, the Commission. The Commission maintains an Internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the Commission at www.sec.gov. Additionally, from time to time, we provide notifications of material news including press releases and conferences on our website. Webcasts of presentations made by our company at certain conferences may also be available from time to time on our website, to the extent the webcasts are available. The content of our website is not intended to be incorporated by reference into this report or in any other report or document we file and any references to these websites are intended to be inactive textual references only. Interested persons may sign up on our website to automatically receive e-mail alerts when we post financial information and issue press releases, and to receive information about upcoming events.
Our website also includes printable versions of our Code of Business Conduct and Ethics and the charters for each of the Audit, Compensation and Nominating Committees of our Board of Directors. Each of these documents is also available in print, free of charge, to any stockholder who requests a copy by addressing a request to:
Protalix BioTherapeutics, Inc.
2 University Plaza, Suite 100
Hackensack, NJ 07601
Attn: Mr. Eyal Rubin, Sr. Vice President and Chief Financial Officer
32
Item 1A. Risk Factors
You should carefully consider the risks described below together with the other information included in this Annual Report on Form 10-K. Our business, financial condition and results of operations could be adversely affected by any of these risks. The risks described below include forward-looking statements, and actual events and our actual results may differ materially from these forward-looking statements. Additional risks and uncertainties not currently known to us or that we currently deem immaterial may also impair our business, results of operations and financial condition. Furthermore, additional risks and uncertainties are described under other captions in this Annual Report on Form 10-K. If any of these risks occur, the value of our common stock could decline. A summary of the risk factors included in this Item 1A are set forth below followed by a full set of risk factors described in greater detail. For further details on our forward-looking statements, see “Cautionary Statement Regarding Forward-Looking Statements” on page 1.
Summary of Risk Factors
Risks Related to the Commercialization and Continued Approval of our Products
There may be safety issues regarding our products that were not known at the time of approval are discovered.
Risks Related to Our Business
Risks Related to Clinical Trials and Regulatory Matters
33
Risks Related to our Financial Condition and Capital Requirements
Risks Related to Intellectual Property Matters
Risks Relating to our Operations in Israel
Risks Related to Investing in our Common Stock
***
34
Risks Related to the Commercialization and Continued Approval of our Products
We currently depend heavily on the generation of revenues from the sales of Elfabrio and Elelyso. Any failure to commercialize Elfabrio, or the experience of significant delays in doing so, will have a material adverse effect on our business, results of operations and financial condition.
We have invested a significant portion of our efforts and financial resources in the development of Elfabrio, and in the past, on the development of Elelyso. Our ability to generate significant product revenues in the future from sales of Elfabrio depends on Chiesi’s successful commercialization of Elfabrio, which we do not control and may not be able to effectively influence, and on the actions and decisions of foreign regulatory authorities. Chiesi may experience delays in, or be unable to achieve, the commercial introduction of Elfabrio globally. Similarly, we generate revenues from sales of Elelyso which are depend on Pfizer’s efforts (except in Brazil), which we do not control. Sales of BioManguinhos alfataliglicerase in Brazil are controlled by Fiocruz. The successful worldwide commercialization of Elfabrio and Elelyso depends on several factors, including the following:
Any failure to commercialize Elfabrio or Elelyso globally or the experience of significant delays in doing so will have a material adverse effect on our business, results of operations and financial condition.
Any current products or future product candidates may cause serious adverse events, or SAEs, undesirable side effects or have other properties that could halt their clinical development, prevent, delay, or cause the withdrawal of their regulatory approval, limit their commercial potential, or result in material negative consequences.
As with most infused products, use of our products or any current or future product candidates could be associated with undesirable side effects or adverse events which can vary in severity and frequency. From time to time, we have observed serious adverse events in clinical studies of our products and product candidates. For example, while conducting clinical trials of Elfabrio, we have observed treatable anaphylactic reactions. Specifically with respect to Elfabrio, as indicated in the boxed warning with which it was approved by the FDA, patients treated with Elfabrio have experienced hypersensitivity reactions, including anaphylaxis. There are postmarketing requirements under the FFDCA included with the approval of Elfabrio by the FDA. For example, the FDA requires that a worldwide descriptive study be conducted that collects prospective and retrospective data in women and their offspring exposed to Elfabrio during pregnancy and/or lactation to assess risk of pregnancy and material complications, adverse effects on the developing fetus and neonate, and adverse effects on the infant. Furthermore, the approval of pharmaceutical products generally includes requirements related to the preparation of a Risk Evaluation and Mitigation Strategy, or REMS, or a risk
35
management plan as required by the EMA, which could include a medication guide outlining the risks of such side effects for distribution to patients, a communication plan for healthcare providers and/or other elements to assure safe use. For example, Chiesi must follow a risk management plan agreed upon with the EMA with respect to Elfabrio, which includes risk minimization measures in connection with risks associated with hypersensitivity reactions and possible medication errors in the home infusion setting.
Our product candidates, if approved, may also be subject to certain post-authorization reporting requirements. As an example, the EMA has required that Chiesi submit pharmacovigilance documents intended to provide post-authorization evaluation of Elfabrio’s risk-benefit balance at defined time points after authorization, beginning within six months of authorization, and that an educational program about home administration be agreed upon with the National Competent Authority prior to the use of Elfabrio in the home setting.
Any of the foregoing, including the boxed warning, could prevent us or our commercialization partners from achieving or maintaining market acceptance of a product or a particular product candidate and could materially harm our business, results of operations, and prospects, and could adversely impact our financial condition, results of operations, ability to raise additional financing or the market price of our common stock.
If safety issues regarding our products that were not known at the time of approval are discovered, or if we or the applicable marketing authorization holder fails to comply with continuing U.S. and applicable foreign regulations, commercialization efforts for our products could be adversely affected and the products could lose their approval or their sales could be suspended.
Drug products remain subject to continuing regulatory oversight after they are approved for marketing, including the review of additional safety information. Drugs are more widely used by patients once approved for sale and, therefore, side-effects and other problems may be observed after approval that were not seen or anticipated, or were not as prevalent or severe, during clinical trials or nonclinical studies. The subsequent discovery of previously unknown problems with a product could negatively affect commercial sales of the product, result in restrictions on the product or lead to the withdrawal of the product from the market. The reporting of adverse safety events involving our products or public speculation about such events could cause our stock price to decline or experience periods of volatility and may have a material adverse effect on our business, results of operations and financial condition.
If we or the marketing authorization holder, or MAH, of any of our products fail to comply with applicable continuing regulatory requirements, we or such MAH may be subject to fines and/or criminal prosecutions, and the product may become subject to suspension or withdrawal of regulatory approval, product recalls and seizures and operating restrictions. In addition, the manufacturers we or an MAH engage to produce a product and the manufacturing facilities in which the product is made are subject to periodic review and inspection by the FDA and foreign regulatory authorities. If problems are identified during the review or inspection of these manufacturers or manufacturing facilities, it could result in the facility becoming unable to manufacture the product or a determination that inventories are not safe for commercial sale, which may have a material adverse effect on our business, results of operations and financial condition.
If physicians, patients, third party payors and others in the medical community do not accept and use Elfabrio, Elelyso or any other future products, our ability to generate revenue from product sales will be materially impaired.
Physicians and patients, and other healthcare providers, may not accept and use Elfabrio, Elelyso or any other future of our other product candidates. Future acceptance and use of any of our products will depend upon a number of factors including:
| ● | perceptions by physicians, patients, third party payors and others in the medical community about the safety and effectiveness of Elfabrio or Elelyso, or any future product, if any; |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
36
| ● | the prevalence and severity of any side effects, including any limitations or warnings contained in our products’ approved labeling, including the boxed warning associated with Elfabrio in the United States; |
| ● | pharmacological benefits of Elfabrio or Elelyso, or any future product, if any relative to competing products and products under development; |
| ● | the efficacy and potential advantages relative to competing products and products under development; |
| ● | relative convenience and ease of administration; |
| ● | effectiveness of education, marketing and distribution efforts by Chiesi, Pfizer and any other relevant licensees and distributors; |
| ● | publicity concerning Elfabrio or Elelyso, or any future product, if any, or concerning competing products and treatments; and |
| ● | the price for our products and competing products. |
If the market opportunities for Elfabrio, Elelyso or any other future products are smaller than we believe they are, our revenues may be adversely affected and our business may suffer.
To date, our development efforts have focused mainly on relatively rare disorders with relatively small patient populations, in particular Gaucher disease and Fabry disease. Estimation of the prevalence of these diseases are based on studies of small subsets of the population of specific geographic areas and are often inexact and prone to error. As new studies are performed, the estimated prevalence of these diseases may change. There can be no assurance that the prevalence of Gaucher disease or Fabry disease in the study populations, particularly in these newer studies, accurately reflect the prevalence of these diseases in the broader world population. If the market opportunities for our products or product candidates are smaller than we believe they are, our revenues may be adversely affected and our business may suffer.
Coverage and reimbursement may not be available for Elfabrio, Elelyso or any other future products in all territories, which could diminish sales of our products or adversely affect the profitably of such sales.
Market acceptance and sales of Elfabrio, Elelyso or any other future products, if any, will depend on coverage and reimbursement policies in the countries in which they are approved for sale. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs they will pay for and establish reimbursement levels. Obtaining reimbursement approval for an approved product from individual governments and other third party payors is a time consuming (six to 12 months or longer) and costly process that requires our collaborators or us, as the case may be, to provide supporting scientific, clinical and cost-effectiveness data for the use of our products, if and when approved, to every payor. Data sufficient to gain acceptance with respect to coverage and reimbursement might not be available, or post-marketing studies may be required in order to demonstrate the cost-effectiveness of approved products, if any, to such payors’ satisfaction. Such studies might require our collaborators or us to commit a significant amount of management time and financial and other resources.
Even if a payor determines that an approved product is eligible for reimbursement, the payor may impose coverage limitations that preclude payment for some uses that are approved by the FDA or other regulatory authorities. For example, the U.S. Inflation Reduction Act allows Medicare to negotiate the prices of the prescription drugs. Such initiatives and legislation may cause added pricing pressure on our products. Proposals that could impact coverage and reimbursement of our products, including giving states more flexibility to manage drugs covered under the Medicaid program and permitting the re-importation of prescription medications from other countries, could have a material adverse effect by limiting our products’ use and coverage. To the extent that private insurers or managed care programs in the United States or elsewhere follow Medicaid coverage and payment developments, they could use the enactment of these increased rebates to exert pricing pressure on our products, and the adverse effects may be magnified by their adoption of lower payment schedules. In addition, full reimbursement may not be available for high priced products. Moreover, eligibility for coverage does not imply that any approved product will be reimbursed in all cases or at a rate
37
that allows us to make a profit or even cover our costs. Limited reimbursement amounts may reduce the demand for, or the price of, Elfabrio, Elelyso or any other future products. If coverage and reimbursement are not available or are available only to limited levels, the sales of Elfabrio, Elelyso or other future products, if any, may be diminished or we may not be able to sell such products profitably.
The pricing of our products in different countries may vary widely, thus creating the potential for third-party trade in our products in an attempt to exploit price differences between countries. This third-party trade of our products could undermine our sales in markets with higher prices which could have a material adverse effect on our business, results of operations and financial condition.
Fiocruz is not complying, and we expect they will continue to not comply, with the terms and conditions of the Brazil Agreement.
We do not control and may not be able to effectively influence Fiocruz’s ability to distribute BioManguinhos alfataliglicerase in Brazil. Fiocruz has not complied with the purchase requirements of the Brazil Agreement in the past, and we expect Fiocruz will continue to not comply and may otherwise materially breach the agreement. Continued non-compliance may have a material adverse effect on our business, results of operations and financial condition.
We face the risk of lower than anticipated purchases of BioManguinhos alfataliglicerase by the Brazilian MoH. In addition, we may fail to supply the intended amounts on time, if at all. We also cannot accurately predict the amount of revenues we will generate under the Brazil Agreement in future periods, if any. Any failure by the Brazilian MoH to purchase BioManguinhos alfataliglicerase, by us to supply BioManguinhos alfataliglicerase for purchase or by Fiocruz to distribute BioManguinhos alfataliglicerase in Brazil, or the experience of significant delays in any of the foregoing, may have a material adverse effect on our business, results of operations and financial condition.
We and our collaborating partners may be subject, directly or indirectly, to federal and state healthcare fraud and abuse and false claims laws and regulations. If we or our collaborating partners are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
All marketing activities associated with products that are approved for sale in the United States, if any, will be, directly or indirectly through our customers, subject to numerous federal and state laws governing the marketing and promotion of pharmaceutical products in the United States, including, without limitation, the federal Anti-Kickback Law, the federal False Claims Act and HIPAA. These laws may adversely impact, among other things, our proposed sales, marketing and education programs.
The federal Anti-Kickback Law prohibits persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce either the referral of an individual, or the furnishing, recommending, or arranging for a good or service, for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value, including for example, gifts, discounts, the furnishing of supplies or equipment, credit arrangements, payments of cash, waivers of co-payments and deductibles, ownership interests and providing anything at less than its fair market value. Despite a series of narrow safe harbors, the federal Anti-Kickback Law prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Penalties for violations of the federal Anti-Kickback Law include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other state or federal healthcare programs. Many states have also adopted laws similar to the federal Anti-Kickback Law, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs, and do not contain identical safe harbors.
The federal False Claims Act imposes liability on any person who, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. In addition, various states have enacted false claims laws analogous to the False Claims Act. Many of these state laws apply where a claim is submitted to any third-party payer and not merely a federal healthcare program. Violations of the federal False Claims Act and the analogous state laws may result in substantial financial penalties, some as much as three times the actual damages sustained by the government.
38
HIPAA created several new federal crimes, including health care fraud, and false statements relating to health care matters. The health care fraud statute prohibits knowingly and willfully executing a scheme to defraud any health care benefit program, including private third-party payers. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services.
We are unable to predict whether we could be subject to actions under any of these or other fraud and abuse laws, or the impact of such actions. Moreover, to the extent that Elfabrio, Elelyso or any other future products, if any, are sold in a foreign country, we and our collaborators may be subject to similar foreign laws and regulations. If we or any of our collaborators are found to be in violation of any of the laws described above and other applicable state and federal fraud and abuse laws, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from government healthcare reimbursement programs and the curtailment or restructuring or our operations, any of which could have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Our Business
We have a limited commercial operating history which may limit the ability of investors to make an informed investment decision.
Elfabrio and Elelyso are our only commercial products both of which are marketed by our commercialization partners. Our operations to date have been limited to acquiring, developing and securing our proprietary technology and undertaking, through third parties, preclinical and clinical trials of our drug candidates and advancing our drug candidates through the regulatory approval processes. These operations provide a limited basis for investors to assess our ability to commercialize our drug candidates and whether to invest in our company.
Any failure by us to supply drug substance to Chiesi or Pfizer may have a material adverse effect on our business, results of operations and financial condition.
We have agreed to sell drug substance to Pfizer and Chiesi for the production of Elelyso and Elfabrio. With respect to Elelyso, our drug substance supply commitment is for a 15-year period after the execution of the Amended Pfizer Agreement, subject to certain terms and conditions. As part of that obligation, we agreed to substantial financial penalties if we fail to comply with the supply commitments, or are delayed in doing so. The amounts of the penalties depend on when any such failure occurs and for how long it persists, if at all, and other considerations. Any failure to comply with the supply commitments to Pfizer and/or Chiesi may have a material adverse effect on our business, results of operations and financial condition.
Our strategy, in certain cases, is to enter into collaboration agreements with third parties to leverage our ProCellEx system to develop product candidates. Failure to enter into such agreements, or non-compliance by us or our collaborators with such agreements, may have a material adverse effect on our business, results of operations and financial condition.
Our strategy, in certain cases, is to enter into arrangements with pharmaceutical companies to leverage our ProCellEx system to develop additional product candidates. Our future revenues may depend, in part, on our ability to enter into and maintain arrangements with our existing partners and other companies having sales, marketing and distribution capabilities and the ability of such companies to successfully market and pharmaceutical products on a global scale. Under these arrangements, we may grant to our partners rights to license and commercialize pharmaceutical products developed under the applicable agreements, as we have done with Elfabrio and Elelyso. Commercialization, marketing, distribution and other similar alliances with respect to our products and product candidates will subject us to a number of risks. We may be required to relinquish important rights to our products or product candidates, and the rights of our partners may limit our flexibility in considering alternatives for the commercialization of our products and product candidates. Our partners may control key decisions relating to the development of the products and we may depend on our partners’ expertise and dedication of sufficient time and resources to develop and commercialize our products and product candidates. Our partners may experience financial difficulties which adversely affect their efforts with respect to our product and product candidates. If we or any of our current or future partners breach or terminate the agreements that make up such arrangements, our partners otherwise fail to conduct their obligations under such arrangements in a timely
39
manner, there is a dispute about their obligations or if either party terminates the applicable agreement or elects not to continue the arrangement, we may not enjoy the benefits of the agreements or receive a sufficient amount of royalty or milestone payments from them, if any, which may have a material adverse effect on our business, results of operations and financial condition.
If we are unable to enhance our portfolio of product candidates, our business may be adversely affected.
A key element of our business strategy is to establish a portfolio of product candidates as targets for development and eventual commercialization. We seek to do so through our internal research programs and strategic collaborations. Research programs to identify new product candidates require substantial technical, financial and human resources, whether or not any product candidates are ultimately identified. A research program may initially show promise in identifying a potential product candidate, yet fail to immediately yield the product candidate for clinical development for many reasons, including the following:
| ● | a product candidate is not capable of being produced in commercial quantities at an acceptable cost, or at all; |
| ● | the research methodology used may not be successful in identifying potential product candidates; or |
| ● | a product candidate may, after further study, be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory approval. |
Any failure or delay in the establishment of a portfolio of product candidates as targets for development and eventual commercialization may have a material adverse effect on our business, results of operations and financial condition.
The manufacture of our products is an exacting and complex process, and any manufacturing problems encountered by us or certain of our providers may have a material adverse effect on our business, results of operations and financial condition.
The FDA and foreign regulators require manufacturers to register manufacturing facilities. The FDA and foreign regulators also inspect these facilities to confirm compliance with cGMP or similar requirements that the FDA or foreign regulators establish. We or certain of our services and materials providers, including our fill and finish service providers, may face manufacturing or quality control problems causing product production and shipment delays or a situation where we or the provider may not be able to maintain compliance with the FDA’s cGMP requirements, or those of foreign regulators, necessary to continue manufacturing. We or any such third-party manufacturer might be unable to formulate and manufacture our products in the volume and of the quality required to meet our preclinical, clinical and commercial needs. If we engage any contract manufacturers, such manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our preclinical, clinical or commercial needs. In addition, we and contract manufacturers are subject to the rules and regulations of the FDA and comparable foreign regulatory authorities and face the risk that any of those authorities may find that they are not in compliance with applicable regulations. To date, our current facility has passed audits by the FDA and a number of other regulatory authorities but remains subject to audit by other foreign regulatory authorities. There can be no assurance that we or our contract manufacturers will be able to comply, or continue to comply, with FDA or foreign regulatory manufacturing requirements, and the failure to so comply, or continue to comply, may have a material adverse effect on our business, results of operations and financial condition.
We rely on third parties for final processing of Elfabrio, Elelyso and our other product candidates, which exposes us to a number of risks that may delay development, regulatory approval and commercialization of Elfabrio, Elelyso or our other product candidates or result in higher product costs.
We have no experience in the final filling and freeze drying steps of the drug manufacturing process. We rely on third-party service providers in the United States and Europe to perform fill and finish activities for Elelyso and Elfabrio, and have engaged other parties for our other product candidates. The number of potential fill and finish services providers is limited and we face the risk of being unable to identify manufacturers and/or replacement manufacturers on acceptable
40
terms or at all. In addition, the FDA and other regulatory authorities, as applicable, must approve any manufacturer and/or replacement manufacturer, including us.
Any failure to identify and maintain fill and finish service providers could delay our preclinical and clinical trials, the approval, if any, of our potential drug candidates by the FDA and other regulatory authorities, or the commercialization of our drug candidates, or could result in higher product costs or otherwise deprive us of potential product revenues.
We have limited experience in selling, marketing or distributing products and limited internal capability to do so.
We currently have very limited sales, marketing or distribution capabilities and no experience in building a sales force and distribution capabilities. Under our arrangements with Pfizer and Chiesi, we have out-licensed the marketing rights to Elfabrio and Elelyso, except that we retained the marketing rights to BioManguinhos alfataliglicerase in Brazil. The commercialization of a product requires the commitment of significant financial and managerial resources to develop a marketing and sales force with technical expertise and with supporting distribution capabilities. If we elect to commercialize our products directly and without strategic partners we may be unable to recruit and retain adequate numbers of effective sales and marketing personnel. In addition, such sales personnel might not access an adequate number of physicians or persuade them to prescribe our products, or may lack complementary products to offer to such physicians. Commercialization by such sales personnel may expose our company to unforeseen costs and expenses.
Developments by competitors may render our products or technologies obsolete or non-competitive which would have a material adverse effect on our business, results of operations and financial condition.
We compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Our products compete, and our products candidates will have to compete, with existing therapies and therapies under development by our competitors. Our commercial opportunities may be reduced or eliminated if our competitors develop and market products that are less expensive, more effective or safer than our products. Other companies have drug candidates in various stages of preclinical or clinical development to treat diseases for which we are also seeking to develop products. Some of these potential competing drugs are further advanced in development than our drug candidates and may be commercialized earlier. See “Business – Competition.”
Most of our competitors, either alone or together with their collaborative partners, operate larger research and development programs, staff and facilities and have substantially greater financial resources than we do, as well as significantly greater experience in:
| ● | developing drugs; |
| ● | undertaking preclinical testing and human clinical trials; |
| ● | obtaining marketing approvals from the FDA and other regulatory authorities; |
| ● | formulating and manufacturing drugs; and |
| ● | launching, marketing and selling drugs. |
These organizations also compete with us to attract qualified personnel, acquisitions and joint ventures candidates and for other collaborations. Activities of our competitors may impose unanticipated costs on our business or adversely affect the market for our products which would have a material adverse effect on our business, results of operations and financial condition.
If we in-license drug candidates, we may delay or otherwise adversely affect the development of our existing drug candidates, which may negatively impact our business, results of operations and financial condition.
In addition to our own internally developed drug candidates, we proactively seek opportunities to in-license and advance other drug candidates that are strategic and have value-creating potential to take advantage of our development know-how and technology. In-licensing additional drug candidates may significantly increase our capital requirements, and
41
place a strain on the time of our existing personnel, which may delay or otherwise adversely affect the development of our existing drug candidates or cause us to re-prioritize our drug pipeline if we do not have the necessary capital resources to develop all of our drug candidates, which may have a material adverse effect on our business, results of operations and financial condition.
If we acquire companies, products or technologies, we may face integration risks and costs associated with those acquisitions that could potentially negatively impact our business, results of operations and financial condition.
If we are presented with appropriate opportunities, we may acquire or make investments in complementary companies, products or technologies. If we acquire companies or technologies, we will face risks, uncertainties and disruptions associated with the integration process, including difficulties in the integration of the operations of an acquired company, integration of acquired technology with our products, diversion of our management’s attention from other business concerns, the potential loss of key employees or customers of the acquired business and impairment charges if future acquisitions are not as successful as we originally anticipate. In addition, our operating results may suffer because of acquisition-related costs or amortization expenses or charges relating to acquired intangible assets. Any failure to successfully integrate other companies, products or technologies that we may acquire may have a material adverse effect on our business, results of operations and financial condition.
We depend upon key employees and consultants in a competitive market for skilled personnel. If we are unable to attract and retain key personnel, it could adversely affect our ability to develop and market our products.
We are highly dependent upon the principal members of our management team, especially our President and Chief Executive Officer, Dror Bashan, as well as the Chairman of our Board of Directors, Eliot Forster, our other directors, consultants and collaborating scientists. Many of these people have been involved with us for many years and have played integral roles in our progress, and we believe that they will continue to provide value to us. A loss of any of these personnel may have a material adverse effect on aspects of our business, clinical development and regulatory programs. We have employment agreements with Mr. Bashan and our other executive officers that may be terminated by us or the applicable officer at any time with varying notice periods of 30 to 180 days. The loss of any of these persons’ services may adversely affect our ability to develop and market our products and obtain necessary regulatory approvals. Further, we do not maintain key-man life insurance.
We also depend in part on the continued service of our key scientific personnel and our ability to identify, hire and retain additional personnel. We experience intense competition for qualified personnel, and the existence of non-competition agreements between prospective employees and their former employers may prevent us from hiring those individuals or subject us to suit from their former employers. While we attempt to provide competitive compensation packages to attract and retain key personnel, many of our competitors are likely to have greater resources and more experience than we have, making it difficult for us to compete successfully for key personnel.
Under current U.S. and Israeli laws, we may not be able to enforce employees’ covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees.
We have entered into non-competition agreements with substantially all of our employees. These agreements prohibit our employees, if they cease working for us, from competing directly with us or working for our competitors for a limited period. Under current U.S. and Israeli laws, we may be unable to enforce these agreements against most of our employees and it may be difficult for us to restrict our competitors from gaining the expertise our former employees acquired while working for us. If we cannot enforce our employees’ non-compete agreements, we may be unable to prevent our competitors from benefiting from the expertise of our former employees, which may have a material adverse effect on our business, results of operations and financial condition.
42
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, stockholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our common stock.
Effective internal control over financial reporting is necessary for us to provide reliable financial reports. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. While our assessment of our internal control over financial reporting resulted in our conclusion that as of December 31, 2023, our internal control over financial reporting was effective, we cannot predict the outcome of our testing or any subsequent testing by our auditor in future periods. Any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act, or Section 404, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal control over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. Inferior internal controls could also cause investors to lose confidence in our reported financial information and affect our reputation, which could have an adverse effect on the trading price of our common stock.
Our management is required to assess the effectiveness of our internal controls and procedures and disclose changes in these controls on an annual basis. However, for as long as we are a non-accelerated filer, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404. An independent assessment of the effectiveness of our internal control could identify deficiencies in internal control over financial reporting that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
Our internal computer systems, or those used by our third-party contractors or consultants, may fail or suffer security breaches, resulting in liability and harm to our reputation, which could negatively affect our business, results of operation and financial condition. We may face liability if we breach our obligations related to the protection, security, nondisclosure of confidential information or disclosure of sensitive data or fail or are perceived to fail to comply with applicable data protection laws and regulations, or consumer protection laws, regulations and standards.
Despite the implementation of security measures, our internal computer systems and those of our present and future contractors and consultants are vulnerable to damage from computer viruses and unauthorized access. Although to our knowledge we have not experienced any material system failure or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on our third-party research institution collaborators for research and development of our product candidates and other third parties for the manufacture of our product candidates and to conduct clinical trials, and similar events relating to their computer systems could also have a material adverse effect on our business. We have a cybersecurity insurance policy to protect us from such risks. However, to the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability despite our insurance policy, and the further development and commercialization of our product candidates could be delayed.
Further, the global data protection landscape is rapidly evolving, and we are or may become subject to numerous local and foreign laws, requirements and regulations governing the collection, use, disclosure, retention, and security of personal data, such as information that we may collect about individuals worldwide. Implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future, and we cannot yet determine the impact future laws, regulations, standards, or perception of their requirements may have on our business. This evolution may create uncertainty in our business or to collect, store, transfer use and share personal information, necessitate the acceptance of more onerous obligations in our contracts, result in liability or impose additional costs on us. The cost of compliance with these laws, regulations and standards is high and is likely to increase in the future. Any failure or perceived failure by us to comply with local or foreign laws or regulation, our internal policies and procedures or our contracts governing our processing of personal information could result in negative publicity, government investigations
43
and enforcement actions, claims by third parties and damage to our reputation, any of which could have a material adverse effect on our business, results of operation and financial condition.
Our failure to adhere to or successfully implement processes in response to changing regulatory requirements in this area could result in legal liability or impairment to our reputation in the marketplace, which could have a material adverse effect on our business, financial condition and results of operations.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and divert management’s attention and resources, which could have a material adverse effect on our business, results of operation and financial condition.
If product liability claims are brought against us, it may result in reduced demand for our products and product candidates or damages that exceed our insurance coverage.
The use and marketing of our products and product candidates, including in connection with clinical trials, exposes us to product liability or similar claims if the use or misuse of those products or product candidates cause injury or disease, or results in adverse effects. We presently carry product liability and clinical trial liability insurance with coverages of up to $10.0 million per occurrence and $10.0 million in the aggregate, an amount we consider reasonable and customary. However, this insurance coverage includes various deductibles, limitations and exclusions from coverage, and in any event might not fully cover any potential claims. In the future, we may need to obtain additional product liability and clinical trial liability coverage; however, such insurance is expensive and insurance companies may not issue this type of insurance when we need it. We may not be able to obtain adequate insurance in the future at an acceptable cost. Any product liability claim, even one that was not in excess of our insurance coverage or one that is meritless and/or unsuccessful, may adversely affect the availability of funds for other purposes, such as research and development, which may have a material adverse effect on our business, results of operations and financial condition. Product liability claims, even if without merit, may result in reduced demand for our products, if approved, or result in adverse market reactions, which would have a material adverse effect on our business, results of operations and financial condition.
Our ability to utilize net operating loss carryforwards may be limited.
Our NOL carryforwards as of December 31, 2023, are equal to approximately $234.8 million, of which approximately $24.4 million may be restricted under Section 382 of the Internal Revenue Code, or the IRC. IRC Section 382 applies whenever a corporation with NOLs experiences an ownership change. As a result of IRC Section 382, the taxable income for any post-change year that may be offset by a pre-change NOL may not exceed the fair market value of the pre-change entity multiplied by the IRC long-term tax exempt rate. Significant judgment is required in determining any valuation allowance recorded against deferred tax assets. In assessing the need for a valuation allowance, we considered all available evidence, including past operating results, the most recent projections for taxable income and prudent and feasible tax planning strategies. We reassess our valuation allowance periodically and if future evidence allows for a partial or full release of the valuation allowance, a tax benefit will be recorded accordingly. Any ownership change (including as a result of conversion of our outstanding convertible notes into shares of our common stock), or any other limitation on our utilization of NOLs, could have a material adverse effect on our business, results of operations and financial condition.
Our corporate structure may create U.S. federal income tax inefficiencies.
Protalix Ltd. is our wholly-owned subsidiary and thus a controlled foreign corporation of our company for U.S. federal income tax purposes. This organizational structure may create inefficiencies, as certain types of income and investments of Protalix Ltd. that otherwise would not be currently taxable under general U.S. federal income tax principles may become taxable. These inefficiencies may require us to use more of our NOLs than we otherwise might and may result in a tax liability without a corresponding distribution from our subsidiary which could have a material adverse effect on our business, results of operations and financial condition.
44
In addition, on December 22, 2017, the U.S.Tax Cuts and Jobs Act, or the TCJA, that significantly reforms the IRC was enacted. The TCJA, among other things, includes changes to U.S. federal tax rates, imposes significant additional limitations on the deductibility of certain expenses and adds certain limitations to the use of net operating loss carryforwards arising after December 31, 2017. Effective in 2022, the TCJA requires all U.S. companies to capitalize and subsequently amortize R&D expenses that fall within the scope of Section 174 over five years for research activities conducted in the United States and over 15 years for research activities conducted outside of the United States rather than deducting such costs in the year incurred for tax purposes. As a result of the TCJA, we expect to incur an increase in our tax expenses and a decrease in our cash flows provided by operations.
We are a holding company with no operations of our own.
We are a holding company with no operations of our own. Accordingly, our ability to conduct our operations, service any current or future debt and pay dividends, if any, is dependent upon the earnings from the business conducted by Protalix Ltd. The distribution of those earnings or advances or other distributions of funds by our subsidiary to us, as well as our receipt of such funds, are contingent upon the earnings of Protalix Ltd. and are subject to various business considerations and U.S. and Israeli laws. If Protalix Ltd. is unable to make sufficient distributions or advances to us, or if there are limitations on our ability to receive such distributions or advances, we may not have the cash resources necessary to conduct our corporate operations or service our debt which would have a material adverse effect on our business, results of operations and financial condition.
Outbreaks of contagious disease or similar public health threats could materially and adversely affect our business, results of operations and financial condition.
Outbreaks of contagious disease or other adverse public health developments worldwide could have a material adverse effect on our business, financial condition and results of operations. Outbreaks of contagious disease or other adverse public health developments could affect our business in a number of ways, including but not limited to:
| ● | Disruptions or restrictions on our employees’ ability to work effectively due to illness; |
| ● | Temporary closures or disruptions at our facilities or the facilities of our contract manufacturers or suppliers could adversely affect our ability to timely meet our customer’s orders and negatively impact our supply chain; |
| ● | Outbreaks of contagious disease could cause delays or disruptions in our supply chain; |
| ● | The failure of third parties on which we rely to meet their respective obligations to us, or significant disruptions in their ability to do so, which may be caused by their own financial or operational difficulties, could have an adverse impact on our business, financial condition or results of operations; and |
| ● | The impact of contagious disease or other adverse public health developments could also exacerbate other risks discussed elsewhere in these Risk Factors, any of which could have a material adverse effect on us. |
Our operating costs and business operations could be adversely affected by climate-related events and increasing regulatory requirements and security.
The effects of climate change (such as drought, flooding, heat waves, wildfires, increased storm severity, and sea level rise, etc.) could affect our ability to continue our operations and cause delays in the development of our existing drug candidates, manufacturing and shipment, all of which could cause reputational harm or otherwise have an adverse effect on our business, results of operation and financial condition. In addition, the impacts of climate change on the global economy and our industry are rapidly evolving. Changing market dynamics, global policy developments and the increasing frequency and impact of extreme weather events on critical infrastructure across different countries could have the potential to disrupt our business, the business of our third-party suppliers and the business of our customers, and may cause us to experience higher attrition, losses and additional costs to maintain or resume operations. We also expect to face increasing regulatory requirements and regulatory scrutiny related to climate matters, resulting in higher associated compliance costs. Failure to uphold, meet or make timely forward progress against our public commitments
45
and goals related to climate action could adversely affect our reputation with suppliers and customers, financial performance or the ability to recruit and retain talent.
Risks Related to Clinical Trials and Regulatory Matters
We may not obtain the necessary U.S., EMA or other worldwide regulatory approvals to commercialize our drug candidates in a timely manner, if at all, which would have a material adverse effect on our business, results of operations and financial condition.
To commercialize our drug candidates worldwide, we need FDA approval, EMA approval and approvals from other countries’ regulators to commercialize our drug candidates elsewhere, as applicable. In order to obtain FDA approval of any of our drug candidates, we must submit to the FDA a BLA or an NDA demonstrating that the drug candidate is safe and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as preclinical studies, as well as human tests, which are referred to as clinical trials. In the European Union, we must submit an MAA to the EMA. Satisfaction of the regulatory requirements of the FDA, EMA and other countries’ regulatory authorities typically takes many years, depends upon the type, complexity and novelty of the drug candidate and requires substantial resources for research, development and testing. The results of clinical trials of our product candidates may fail to demonstrate that the candidates are safe and effective for their intended uses.
Companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced or late-stage clinical trials, even after obtaining promising earlier trial results or in preliminary findings or other comparable authorities for such clinical trials. Further, even if favorable testing data is generated by the clinical trials of a drug candidate, the applicable regulatory authority may not accept or approve a marketing application we file for the drug candidate or may require us to conduct additional clinical testing or perform post-marketing studies which would cause us to incur additional costs.
Failure to obtain approval of the FDA, EMA or comparable foreign authorities of any of our product candidates in a timely manner, if at all, will severely undermine our business, results of operations and financial condition by decreasing our ability to generate product revenues from the sales of such product candidates.
We are subject to extensive governmental regulation including the requirements of the FDA and other comparable regulatory authorities before our drug candidates may be marketed.
Both before and after marketing approval of our drug candidates, if at all, we, our drug candidates, our suppliers, our contract manufacturers and our contract testing laboratories are subject to extensive regulation by the FDA and comparable foreign regulatory authorities. Failure to comply with applicable requirements of the FDA or comparable foreign regulatory authorities could result in, among other things, any of the following actions:
| ● | warning letters; |
| ● | fines and other monetary penalties; |
| ● | unanticipated expenditures; |
| ● | delays in the FDA’s or other foreign regulatory authorities’ approving, or the refusal of any regulatory authority to approve, any drug candidate; |
| ● | product recall or seizure; |
| ● | interruption of manufacturing or clinical trials; |
| ● | operating restrictions; |
| ● | injunctions; and |
| ● | criminal prosecutions. |
46
In addition to the approval requirements, other numerous and pervasive regulatory requirements apply, both before and after approval, to us, our drug candidates, our suppliers, contract manufacturers, and contract testing laboratories. These include requirements related to:
| ● | testing; |
| ● | manufacturing; |
| ● | quality control; |
| ● | labeling; |
| ● | advertising; |
| ● | promotion; |
| ● | distribution; |
| ● | export; |
| ● | reporting to the FDA certain adverse experiences associated with use of the drug candidate; and |
| ● | obtaining additional approvals for certain modifications to the drug candidate or its labeling or claims. |
We also are subject to inspection by the FDA and comparable foreign regulatory authorities, to determine our compliance with regulatory requirements, as are our suppliers, contract manufacturers, and contract testing laboratories, and there can be no assurance that the FDA, or any other comparable foreign regulatory authority, will not identify compliance issues that may disrupt production or distribution, or require substantial resources to correct. We may be required to make modifications to our manufacturing operations in response to these inspections which may require significant resources and may have a material adverse effect upon our business, results of operations and financial condition.
The approval process for any drug candidate may also be delayed by changes in government regulation, future legislation or administrative action or changes in policy of the FDA and comparable foreign authorities that occur prior to or during their respective regulatory reviews of such drug candidate.
Delays in obtaining regulatory approvals with respect to any drug candidate will materially and adversely affect our prospects, business, results of operations and financial condition.
Delays in obtaining regulatory approvals with respect to any drug candidate may:
| ● | delay commercialization of, and our ability to derive product revenues from, such drug candidate; |
| ● | delay any regulatory-related milestone payments payable under outstanding collaboration agreements; |
| ● | require us to perform costly procedures with respect to such drug candidate; or |
| ● | otherwise diminish any competitive advantages that we may have with respect to such drug candidate. |
Delays in the approval process for any drug candidate may have a material adverse effect upon our prospects, business, results of operations and financial condition.
47
Clinical trials are very expensive, time-consuming and difficult to design and implement and may result in unforeseen costs, which may have a material adverse effect on our business, results of operations and financial condition.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. Preliminary and initial results from a clinical trial do not necessarily predict final results, and failure can occur at any stage of the trial. We may encounter problems that could cause us to abandon or repeat preclinical studies or clinical trials. The clinical trial process is also time-consuming. Failure or delay in the commencement or completion of our clinical trials may be caused by several factors, including:
| ● | slower than expected rates of patient recruitment, particularly with respect to trials of rare diseases; |
| ● | determination of dosing issues; |
| ● | unforeseen safety issues; |
| ● | lack of effectiveness during clinical trials; |
| ● | disagreement by applicable regulatory bodies over our trial protocols, the interpretation of data from preclinical studies or clinical trials or conduct and control of clinical trials; |
| ● | determination that the patient population participating in a clinical trial may not be sufficiently broad or representative to assess efficacy and safety for our target population; |
| ● | inability to monitor patients adequately during or after treatment; |
| ● | inability or unwillingness of medical investigators and institutional review boards to follow our clinical protocols; and |
| ● | lack of sufficient funding to finance the clinical trials. |
Any failure or delay in commencement or completion of any clinical trials of our product candidates will have a material adverse effect on our business, results of operations and financial condition. In addition, we, the FDA or other regulatory authorities may suspend any clinical trial at any time if it appears that we are exposing participants in the trial to unacceptable safety or health risks or if the FDA or such other regulatory authorities, as applicable, find deficiencies in our IND submissions or the conduct of the trial. Any suspension of a clinical trial may have a material adverse effect on our business, results of operations and financial condition.
If the results of our clinical trials do not support our claims relating to a drug candidate, or if serious side effects are identified, the completion of development of such drug candidate may be significantly delayed or we may be forced to abandon development altogether, which will significantly impair our ability to generate product revenues.
The results of our clinical trials with respect to any drug candidate might not support our claims of safety or efficacy, the results of our clinical trials may fail to conclusively show superiority over other commercially available treatments for the same or similar indications, the effects of our drug candidates may not be the desired effects or may include undesirable side effects or the drug candidates may have other unexpected characteristics. Data obtained from tests are susceptible to varying interpretations which may delay, limit or prevent regulatory approval. The clinical trial process may fail to demonstrate that our drug candidates are safe for humans and effective for indicated uses. In addition, our clinical trials, may involve specific and small patient populations. Results of early clinical trials conducted on a small patient population may not be indicative of future results. Adverse or inconclusive results may cause us to abandon a drug candidate and may delay development of other drug candidates. Any delay in, or termination of, our clinical trials will delay the filing of BLAs and NDAs with the FDA, or other filings with other foreign regulatory authorities, and, ultimately, significantly impair our ability to commercialize our drug candidates and generate product revenues which would have a material adverse effect on our business, results of operations and financial condition.
48
Interim, top-line or preliminary data from clinical trials that we announce or publish may change as more patient data becomes available and are subject to audit and verification procedures that could result in material changes in the final data.
We may publicly disclose interim, top-line or preliminary data from our clinical trials, which is based on a preliminary analysis of then-available data. The results and related findings and conclusions are subject to change following a full analysis of all data related to the particular trial. We also may make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, any interim, top-line or preliminary results that we report may differ from future results of the same trials, or different conclusions or considerations may qualify such results once additional data have been received and fully evaluated. Top-line data also remain subject to audit and verification procedures that may result in the final data being materially different than the preliminary data we previously published. As a result, any top-line data should be viewed with caution until final data are available. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more data becomes available. Further, regulatory agencies may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses, or may interpret or ascribe different weight to the data, which may impact the value of the clinical trial and may affect the particular clinical program and the approvability or commercialization of the particular product candidate and our business in general. If regulatory authorities disagree with the conclusions we reach, we may not be able to obtain approval for and commercialize our product candidates, which will have a material adverse effect on our business, results of operations and financial condition.
We may find it difficult to enroll patients in our clinical trials, or patients may discontinue their participation in our clinical trials, which could cause significant delays in the completion of such trials or may negatively impact the results of these studies and extend the timeline for completion of our development programs or cause us to abandon one or more clinical trials.
Some of the diseases or disorders that our drug candidates under evaluation are intended to treat are relatively rare and we expect only a subset of the patients with these diseases to be eligible for our clinical trials. Our clinical trials generally mandate that a patient cannot be involved in another clinical trial for the same indication. Therefore, subjects that participate in ongoing clinical trials for products that are competitive with our drug candidates are not available for our clinical trials. An inability to enroll a sufficient number of patients for any of our clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether. In addition, patients who enroll in our clinical trials may discontinue their participation at any time during the study as a result of a number of factors, related to the trials or not, including withdrawing their consent, experiencing adverse clinical events, which may or may not be judged related to our drug candidates, or due to personal reasons, such as planned or actual pregnancies. The discontinuation of patients in any one of our studies may delay the completion of the study or cause the results from the study not to be positive or to not support a filing for regulatory approval of the applicable drug candidate. Any failure to enroll a sufficient number of patients in our clinical trials in a timely manner, if at all, may have a material adverse effect on our business, results of operations and financial condition.
Our clinical trials depend upon third-party service providers and, accordingly, the results of our clinical trials and such research activities are subject to delays and other risks which are, to a certain extent, beyond our control, which could impair our clinical development programs and our competitive position.
We depend upon independent investigators and collaborators, such as universities and medical institutions, to conduct our preclinical and clinical trials. These collaborators are not our employees, and we cannot control the amount or timing of resources that they devote to our clinical development programs. The investigators may not prioritize to our clinical development programs or pursue them as diligently as we would if we were undertaking such programs directly. If outside collaborators fail to devote sufficient time and resources to our clinical development programs, or if their performance is substandard, the approval of anticipated NDAs, BLAs and other marketing applications, and our introduction of new drugs, if any, may be delayed which could impair our clinical development programs and would have a material adverse effect on our business, results of operations and financial condition. Our collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators also assist our competitors, our competitive position could be harmed.
49
We have only limited experience in regulatory affairs, and some of our drug candidates may be based on new technologies. These factors may affect our ability or the time we require to obtain necessary regulatory approvals.
We have only limited experience in filing and prosecuting the applications necessary to gain regulatory approvals for medical devices and drug candidates. Moreover, some of the drug candidates that are likely to result from our development programs may be based on new technologies that have not been extensively tested in humans. The regulatory requirements governing these types of drug candidates may not be well defined or more rigorous than for conventional products. As a result, we may experience a longer regulatory process in obtaining regulatory approvals of any products that we develop, which may have a material adverse effect on our business, results of operations and financial condition.
We may seek orphan drug designation for some or all of our product candidates across various indications, but we may be unable to obtain such designations or to maintain the benefits associated with orphan drug designation, including market exclusivity, which may cause our revenue, if any, to be reduced.
We may seek orphan drug or other comparable designations for our product candidates in specific indications in which there is a medically plausible basis for the use of these products. Even if we obtain orphan drug designation, exclusive marketing rights in the United States or other applicable jurisdictions may be limited if we seek approval for an indication broader than the orphan designated indication, and may be lost if the FDA, or other applicable regulatory authorities later determine that the request for designation was materially defective, if we are unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition, or if a subsequent applicant demonstrates clinical superiority over our products, if approved. In addition, more than one drug can have orphan designation for the same indication. Although we may seek orphan drug designation for other product candidates, we might not receive such designations.
Risks Related to our Financial Condition and Capital Requirements
Servicing our debt and settling conversion requests may require a significant amount of cash, and we may not have sufficient cash flow from our business to pay our debt. Furthermore, restrictive covenants governing our indebtedness may restrict our ability to raise additional capital.
As of December 31, 2023, we have outstanding $20.4 million aggregate principal amount of our 2024 Notes which are secured with a perfected lien on all of our assets. Under the terms of the indenture governing the 2024 Notes, or the 2024 Indenture, we are required to maintain a minimum cash balance of at least $7.5 million. Our ability to make payments with respect to the 2024 Notes and to satisfy any other debt obligations depends on our future operating performance and our ability to generate significant cash flow in the future, which will be affected by prevailing economic conditions and financial, business, competitive, legislative and regulatory factors as well as other factors affecting our company and industry, many of which are beyond our control. If, when required, we are unable to comply with the terms of the 2024 Notes, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. In addition, certain terms of the 2024 Notes regarding the security interest or future indebtedness may restrict us from adopting any of these alternatives. We may be unable to obtain amendments and waivers of such restrictions. If there is a default on such notes, the note holders could, among other things, elect to declare all amounts owed immediately due and payable, which could cause all or a large portion of our available cash flow to be used to pay such amounts and thereby reduce the amount of cash available to pursue our business plans or force us into bankruptcy or liquidation, or, with respect to our indebtedness that is secured, result in the foreclosure on the assets that secure the debt, which would force us to relinquish rights to assets that we may believe are critical to our business. Any default on our debt will have a material adverse effect on our business, results of operations and financial condition.
Our significant level of indebtedness could adversely affect our business, results of operations and financial condition and prevent us from fulfilling our obligations under our convertible notes and our other indebtedness.
Our 2024 Notes represent a significant amount of indebtedness with substantial debt service requirements. We may also incur additional indebtedness to meet future financing needs. Our substantial indebtedness could have material adverse effects on our business, results of operations and financial condition. For example, it could:
50
| ● | make it more difficult for us to satisfy our financial obligations, including with respect to the 2024 Notes; |
| ● | result in an event of default under the 2024 Indenture if we fail to comply with the financial and other restrictive covenants contained in agreements governing any future indebtedness, which event of default could result in all of our debt becoming immediately due and payable; |
| ● | increase our vulnerability to general adverse economic, industry and competitive conditions; |
| ● | reduce the availability of our cash flow to fund working capital, capital expenditures, acquisitions and other general corporate purposes because we will be required to dedicate a substantial portion of our cash flow from operations to the payment of principal and interest on our indebtedness; |
| ● | limit our flexibility in planning for, or reacting to, and increasing our vulnerability to changes in our business, the industry in which we operate and the general economy; |
| ● | prevent us from raising funds necessary to purchase 2024 Notes surrendered to us by holders upon a fundamental change (as described in the 2024 Indenture), which failure would result in an event of default with respect to the 2024 Notes; |
| ● | place us at a competitive disadvantage compared to our competitors that have less indebtedness or are less highly leveraged and that, therefore, may be able to take advantage of opportunities that our debt levels or leverage prevent us from exploiting; and |
| ● | limit our ability to obtain additional financing. |
Each of these factors may have a material and adverse effect on our business, results of operations and financial condition and our ability to meet our payment obligations relating to the 2024 Notes and our other indebtedness.
We are required to comply with a number of covenants under the 2024 Indenture governing our outstanding 2024 Notes that could hinder our growth.
The 2024 Indenture contains a number of restrictive affirmative and negative covenants, which limit our ability to incur additional debt; exceed certain limits; pay dividends or distributions; or merge, consolidate or dispose of substantially all of our assets, including all of our intellectual property assets and other material assets securing the 2024 Notes. A breach of these covenants could result in default, and if such default is not cured or waived, the holders of the indebtedness could, among other things, elect to declare all amounts owed immediately due and payable, which could cause all or a large portion of our available cash flow to be used to pay such amounts and thereby reduce the amount of cash available to pursue our business plans or force us into bankruptcy or liquidation, or, result in the foreclosure on the assets that secure the debt, including all of our intellectual property assets, which would force us to relinquish rights to such assets that we may believe are critical to our business. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations. Any default on our debt will have a material adverse effect on our business, results of operations and financial condition.
Any conversion of our outstanding 2024 Notes into common stock will dilute the ownership interest of our existing stockholders, including holders who had previously converted their notes.
The conversion of some or all of our 2024 Notes into shares of our common stock will dilute the ownership interests of our existing stockholders. Any sales in the public market of our common stock issuable upon such conversions could adversely affect prevailing market prices of our common stock. In addition, the existence of our outstanding 2024 Notes may encourage short selling by market participants because the conversion of 2024 Notes could depress the market price of our common stock.
51
The fundamental change purchase feature of our outstanding 2024 Notes may delay or prevent an otherwise beneficial attempt to take over our company.
The terms of our outstanding 2024 Notes require us to offer to purchase the notes for cash in the event of a fundamental change. A non-stock takeover of our company may trigger the requirement that we purchase the notes. This feature may have the effect of delaying or preventing a takeover of our company that would otherwise be beneficial to our stockholders.
We may fail to meet the continued market capitalization-based listing requirement or other continued listing requirements of the NYSE American.
The stock market in general, and the market for pharmaceutical companies in particular, have experienced extreme price and volume fluctuations that may have been unrelated or disproportionate to the operating performance of the listed companies. The trading price of our common stock has been volatile and has been subject to wide price fluctuations in response to various factors, many of which are beyond our control. The volatility of our stock price has from time to time in recent periods affected our market capitalization. Adverse fluctuations in the price per share of our common stock or our market capitalization may result in our failure to meet the continued listing requirements of the NYSE American, which would require us to take steps to gain compliance with alternate listing standards or take remedial steps to bring us into compliance. A failure to maintain or regain compliance with applicable listing standards could adversely affect the liquidity of our common stock and could result in an event of default under the 2024 Indenture, which would have a material adverse effect on our business, results of operations and financial condition.
We may need to raise additional capital to operate our business, which may not be available on favorable terms, or at all, and which will have a dilutive effect on our stockholders.
For the years ended December 31, 2022 and 2021, we had net losses from continuing operations of $14.9 million and $27.6 million, respectively, and, in the year ended December 31, 2023, we had a net income of $8.3 million. The net losses for the years ended December 31, 2022 and 2021 were primarily the result of expenses incurred through a combination of research and development activities and expenses supporting those activities, which includes share-based compensation expense. We may incur losses in future fiscal years as we continue to execute on our strategic goals as drug development and commercialization is very capital intensive. We expect to continue to incur significant operating expenditures, and we anticipate that our expenses will increase in the foreseeable future as we seek to in-license additional technologies, continue to undertake preclinical development and clinical trials for our current and new drug candidates and seek regulatory approvals for our drug candidates. In addition, changes may occur that could consume our existing capital at a faster rate than projected, including, among others, the cost and timing of regulatory approvals, changes in the progress of our research and development efforts and the costs of protecting our intellectual property rights.
Currently, our source of potential revenues will be from royalties and commercial milestone payments generated from Pfizer, Fiocruz and Chiesi. We have also generated revenues from research and development services and milestone and other payments we received in connection with our agreements with our commercialization partners, and generated capital from equity and debt offerings and other sources. The revenues we generate from royalties and commercial milestone payments and other sources may not be sufficient to fund our planned operations and capital expenditures. Accordingly, we may need to finance our future cash needs through corporate collaboration, licensing or similar arrangements, public or private equity offerings or debt financings. If we are unable to secure additional financing in the future on acceptable terms, or at all, we may be unable to expand our portfolio of product candidates, commence or complete planned preclinical and clinical trials or obtain approval of our drug candidates from the FDA and other regulatory authorities. In addition, we may be forced to reduce or discontinue product development or product licensing, reduce or forego sales and marketing efforts and other commercialization activities or forego attractive business opportunities in order to improve our liquidity and to enable us to continue operations which would have a material adverse effect on our business and results of operations. Furthermore, any additional source of financing will likely involve the issuance of our equity securities, which will have a dilutive effect on our stockholders.
52
Risks Related to Intellectual Property Matters
The intellectual property and assets owned by our subsidiaries are subject to security agreements that secure our payment and other obligations under our convertible notes, and our subsidiaries have guaranteed all of those obligations.
In connection with the issuance of our 2024 Notes, we entered into new security agreements pursuant to which our subsidiaries provided first priority security interests in all of their assets, which consist of all of our intellectual property and other material assets. The security agreements secure certain payment, indemnification and other obligations under the 2024 Notes. If we were to default on certain of our obligations, or in certain other circumstances generally related to a bankruptcy or insolvency, holders of our outstanding 2024 Notes could seek to foreclose on the collateral under the security agreements to obtain satisfaction of our obligations, and our business could be materially and adversely impacted, which would in turn have a material adverse effect on our results of operations and financial condition.
Furthermore, in connection with the issuance of the 2024 Notes, our subsidiaries guaranteed all of our obligations under the 2024 Indenture. If we were to default on our obligations under the 2024 Indenture, the holders could require our subsidiaries to satisfy all of those obligations under the guarantees.
If we fail to adequately protect or enforce our intellectual property rights or secure rights to third party patents, the value of our intellectual property rights would diminish and our business, competitive position and results of operations would suffer.
As of December 31, 2023, we had approximately 50 pending patent applications. However, the filing of a patent application does not mean that we will be issued a patent, or that any patent eventually issued will be as broad as requested in the patent application or sufficient to protect our technology. Any modification required to a current patent application may delay the approval of such patent application which may have a material adverse effect on our business, results of operations and financial condition. In addition, there are a number of factors that could cause our patents, if granted, to become invalid or unenforceable or that could cause our patent applications to not be granted, including known or unknown prior art, deficiencies in the patent application or the lack of originality of the technology. Our competitive position and future revenues will depend in part on our ability and the ability of our licensors and collaborators to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. We have filed U.S. and international patent applications for process patents, as well as composition of matter patents, for Elfabrio, Elelyso and our product candidates. However, we cannot predict:
| ● | the degree and range of protection any patents will afford us against competitors and those who infringe upon our patents, including whether third parties will find ways to invalidate or otherwise circumvent our licensed patents; |
| ● | if and when patents will issue; |
| ● | whether or not others will obtain patents claiming aspects similar to those covered by our licensed patents and patent applications; or |
| ● | whether we will need to initiate litigation or administrative proceedings, which may be costly, whether we win or lose. |
As of December 31, 2023, we held, or had license rights to, approximately 90 patents. If patent rights covering our products or technologies are not sufficiently broad, they may not provide us with sufficient proprietary protection or competitive advantages against competitors with similar products and technologies. Furthermore, if the USPTO or foreign patent offices issue patents to us or our licensors, others may challenge the patents or circumvent the patents, or the patent office or the courts may invalidate the patents. Thus, any patents we own or license from or to third parties may not provide any protection against our competitors and those who infringe upon our patents.
53
Furthermore, the life of our patents is limited. The patents we hold, and the patents that may be issued in the future based on patent applications from the patent families, relating to our ProCellEx protein expression system are expected to expire by 2025.
We rely on confidentiality agreements that could be breached and may be difficult to enforce which could have a material adverse effect on our business and competitive position.
Our policy is to enter agreements relating to the non-disclosure of confidential information with third parties, including our contractors, consultants, advisors and research collaborators, as well as agreements that purport to require the disclosure and assignment to us of the rights to the ideas, developments, discoveries and inventions of our employees and consultants while we employ them. However, these agreements can be difficult and costly to enforce. Moreover, to the extent that our contractors, consultants, advisors and research collaborators apply or independently develop intellectual property in connection with any of our projects, disputes may arise as to the proprietary rights to the intellectual property. If a dispute arises, a court may determine that the right belongs to a third party, and enforcement of our rights can be costly and unpredictable. In addition, we rely on trade secrets and proprietary know-how that we seek to protect in part by confidentiality agreements with our employees, contractors, consultants, advisors and others. Despite the protective measures we employ, we still face the risk that:
| ● | these agreements may be breached; |
| ● | these agreements may not provide adequate remedies for the applicable type of breach; or |
| ● | our trade secrets or proprietary know-how will otherwise become known. |
Any breach of our confidentiality agreements or our failure to effectively enforce such agreements may have a material adverse effect on our business and competitive position.
If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages and required to defend against litigation which could result in substantial costs and may have a material adverse effect on our business, results of operations and financial condition.
We have not received to date any claims of infringement by any third parties. However, as our drug candidates progress into clinical trials and commercialization, if at all, our public profile and that of our drug candidates may be raised and generate such claims. Defending against such claims, and occurrence of a judgment adverse to us, could result in unanticipated costs and may have a material adverse effect on our business and competitive position. If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we may incur substantial costs and we may have to:
| ● | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| ● | redesign our products or processes to avoid infringement; |
| ● | stop using the subject matter claimed in the patents held by others, which could cause us to lose the use of one or more of our drug candidates; |
| ● | defend litigation or administrative proceedings that may be costly whether we win or lose, and which could result in a substantial diversion of management resources; or |
| ● | pay damages. |
Any costs incurred in connection with such events or the inability to sell our products may have a material adverse effect on our business, results of operations and financial condition.
54
If we cannot meet requirements under our license agreements, we could lose the rights to our products, which could have a material adverse effect on our business.
We depend on licensing agreements with third parties to maintain the intellectual property rights to certain of our product candidates. Our license agreements require us to make payments and satisfy performance obligations in order to maintain our rights under these agreements. All of these agreements last either throughout the life of the patents that are the subject of the agreements, or with respect to other licensed technology, for a number of years after the first commercial sale of the relevant product.
In addition, we are responsible for the cost of filing and prosecuting certain patent applications and maintaining certain issued patents licensed to us. If we do not meet our obligations under our license agreements in a timely manner, we could lose the rights to our proprietary technology which could have a material adverse effect on our business, results of operations and financial condition.
Risks Relating to our Operations in Israel
Significant parts of our operations are located in Israel and, therefore, our results may be adversely affected by political, economic and military conditions in Israel.
Our executive office and operations are located in the State of Israel. Accordingly, economic, geopolitical and military conditions in Israel and the surrounding region could directly affect our business. Any armed conflicts, political instability, terrorism, cyberattacks or any other hostilities involving Israel or the interruption or curtailment of trade between Israel and its present trading partners could adversely affect our operations. Since the establishment of Israel in 1948, a number of armed conflicts have occurred between Israel and its Arab neighbors, Hamas and Hezbollah. In October 2023, terrorists from the Hamas organization infiltrated Israel’s southern border from the Gaza Strip and in other areas within the State of Israel attacking a number of civilian and military targets while simultaneously launching extensive rocket attacks on the Israeli population and industrial centers. At the same time, clashes between Israel and Hezbollah in Lebanon have increased. In response, Israel’s security cabinet declared war against the Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks. The attacks by Hamas and Hezbollah, and Israel’s defensive measures, as well as actions that could be taken in the future by NATO, the United States, the United Kingdom the European Union or Israel’s neighboring states and other countries have created global security concerns that may result in a greater or lasting regional conflict. To date, our operations have not been adversely affected by this situation. Hostilities have not taken place where our facilities are located and we do not anticipate any disruption to the supply of Elfabrio or Elelyso. However, our facilities are in range of certain of the rockets that are being fired from Lebanon and elsewhere. We suffered minimal damages during a rocket attack in 2006. Our insurance policies do not cover damages incurred in connection with these conflicts or for any resulting disruption in our operations. The Israeli government, as a matter of law, provides coverage for the reinstatement value of direct damages that are caused by terrorist attacks or acts of war; however, the government may cease providing such coverage or the coverage might not be enough to cover potential damages. Any damage to our facilities as a result of war or other hostile action may have a material adverse effect on our business, results of operations and financial condition.
Ongoing and revived hostilities or other Israeli political or economic factors, could prevent or delay shipments of our products, harm our operations and product development and cause any future sales to decrease. If hostilities disrupt the ongoing operation of our facilities or the airports and seaports on which we depend to import and export our supplies, materials, drug substance and other products, our business, results of operations and financial condition may be materially and adversely affected.
Parties with whom we do business may sometimes decline to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary to meet our business partners face to face. Further, shifting economic and political conditions in the United States and in other countries may result in changes in how the United States and other countries conduct business and other relations with Israel, which may have an adverse impact on our Israeli operations and a material adverse impact on our business. In addition, several countries, principally in the Middle East, restrict doing business with Israel, and additional countries may impose restrictions on doing business with Israel and Israeli companies whether as a result of hostilities in the region or otherwise. Moreover, there have been
55
increased efforts by organizations and movements to cause companies and consumers to boycott Israeli goods based on Israeli government policies.
Our operations may be disrupted by the obligations of our personnel to perform military service which could have a material adverse effect on our business.
Many of our male employees in Israel, including members of senior management, are obligated to perform up to one month (in some cases more) of annual military reserve duty until they reach the age of 45 and, in the event of a military conflict, could be called to active duty. To date, we have not suffered any disruptions to our operations from the absence of employees in connection with the current military action. However, we continue to face the risk that our operations could be disrupted by the absence of a significant number of our employees related to military service or the absence for extended periods of military service of one or more of our key employees. Any such disruption may have a material adverse effect on our business, results of operations and financial condition.
A certain portion of our expenses is incurred in New Israeli Shekels and, accordingly, our results of operations may be seriously harmed by currency fluctuations and inflation.
We report our financial statements in U.S. dollars, our functional currency. Although most of our expenses are incurred in U.S. dollars, we pay a portion of our expenses in New Israeli Shekels, or NIS, and as a result, we are exposed to risk to the extent that the inflation rate in Israel exceeds the rate of devaluation of the NIS in relation to the U.S. dollar or if the timing of these devaluations lags behind inflation in Israel. In that event, the U.S. dollar cost of our operations in Israel will increase and our U.S. dollar-measured results of operations will be adversely affected. To the extent that the value of the NIS increases against the U.S. dollar, our expenses on a dollar cost basis increase. Our operations also could be adversely affected if we are unable to guard against currency fluctuations in the future. To date, we have not engaged in hedging transactions. In the future, we may enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rate of the U.S dollar against the NIS. These measures, however, may not adequately protect us from material adverse effects.
The tax benefits available to us require that we meet several conditions and may be terminated or reduced in the future, which would increase our taxes.
We are able to take advantage of tax exemptions and reductions resulting from the “Approved Enterprise” status of our facilities in Israel. To remain eligible for these tax benefits, we must continue to meet certain conditions, including making specified investments in property and equipment, and financing at least 30% of such investments with share capital. If we fail to meet these conditions in the future, the tax benefits would be canceled and we may be required to refund any tax benefits we already have enjoyed. These tax benefits are subject to investment policy by the Investment Center and may not be continued in the future at their current levels or at any level. In recent years the Israeli government has reduced the benefits available and has indicated that it may further reduce or eliminate some of these benefits in the future. The termination or reduction of these tax benefits or our inability to qualify for additional “Approved Enterprise” approvals may increase our tax expenses in the future, which would reduce our expected profits and adversely affect our business and results of operations. Additionally, if we increase our activities outside of Israel, for example, by future acquisitions, such increased activities generally may not be eligible for inclusion in Israeli tax benefit programs.
The Israeli government grants we have received for certain research and development expenditures restrict our ability to manufacture products and transfer technologies outside of Israel and require us to satisfy specified conditions. If we fail to satisfy these conditions, we may be required to refund grants previously received together with interest and penalties which could have a material adverse effect on our business and results of operations.
In the past, our research and development efforts have been financed, in part, through grants that we have received from NATI. We, therefore, must comply with the requirements of the Research Law. Under the Research Law we are prohibited from manufacturing products developed using these grants outside of the State of Israel without special approvals, although the Research Law does enable companies to seek prior approval for conducting manufacturing activities outside of Israel without being subject to increased royalties. We may not receive the required approvals for any proposed transfer of manufacturing activities. Even if we do receive approval to manufacture products developed
56
with government grants outside of Israel, we may be required to pay an increased total amount of royalties (possibly up to 600% of the grant amounts plus interest), depending on the manufacturing volume that is performed outside of Israel, as well as a possibly increased royalty rate. This restriction may impair our ability to outsource manufacturing or engage in similar arrangements for those products or technologies.
Additionally, under the Research Law, Protalix Ltd. is prohibited from transferring NATI-financed technologies and related intellectual property rights outside of the State of Israel, except under limited circumstances and only with the approval of NATI Council or the Research Committee. Protalix Ltd. may not receive the required approvals for any proposed transfer and, if received, Protalix Ltd. may be required to pay NATI a portion of the consideration that it receives upon any sale of such technology by a non-Israeli entity. The scope of the support received, the royalties that Protalix Ltd. has already paid to NATI, the amount of time that has elapsed between the date on which the know-how was transferred and the date on which NATI grants were received and the sale price and the form of transaction will be taken into account in order to calculate the amount of the payment to NATI. Approval of the transfer of technology to residents of the State of Israel is required, and may be granted in specific circumstances only if the recipient abides by the provisions of applicable laws, including the restrictions on the transfer of know-how and the obligation to pay royalties. No assurance can be made that approval to any such transfer, if requested, will be granted.
These restrictions may impair our ability to sell our technology assets or to outsource manufacturing outside of Israel. The restrictions will continue to apply for a certain period of time even after we have repaid the full amount of royalties payable for the grants. If we fail to satisfy the conditions of the Research Law, we may be required to refund certain grants previously received together with interest and penalties, and may become subject to criminal charges, any of which could have a material adverse effect on our business, results of operations and financial condition.
Investors may have difficulties enforcing a U.S. judgment, including judgments based upon the civil liability provisions of the U.S. federal securities laws against us, our executive officers and most of our directors or asserting U.S. securities laws claims in Israel.
A majority of our directors and all of our executive officers are residents of Israel, and accordingly, most of their assets and our assets are located outside the United States. Service of process upon us or our non-U.S. resident directors and officers and enforcement of judgments obtained in the United States against us or our non-U.S. resident directors and executive officers may be difficult to obtain within the United States. We have been informed by our legal counsel in Israel that it may be difficult to assert claims under U.S. securities laws in original actions instituted in Israel or obtain a judgment based on the civil liability provisions of U.S. federal securities laws. Israeli courts may refuse to hear a claim based on a violation of U.S. securities laws against us or our non-U.S. resident officers and directors because Israel may not be the most appropriate forum to bring such a claim. In addition, even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by Israeli law. There is little binding case law in Israel addressing the matters described above. Israeli courts might not enforce judgments rendered outside Israel, which may make it difficult to collect on judgments rendered against us or our non-U.S. resident officers and directors.
Moreover, among other reasons, including but not limited to, fraud or absence of due process, or the existence of a judgment which is at variance with another judgment that was given in the same matter if a suit in the same matter between the same parties was pending before a court or tribunal in Israel, an Israeli court will not enforce a non-Israeli judgment if it was given in a state whose laws do not provide for the enforcement of judgments of Israeli courts (subject to exceptional cases) or if its enforcement is likely to prejudice the sovereignty or security of the State of Israel.
Risks Related to Investing in our Common Stock
The market price of our common stock may fluctuate significantly.
The market price of our common stock has experienced significant volatility. The securities of life sciences companies often experience significant volatility in connection with clinical trial and regulatory announcements.
57
We anticipate that the market price of our common stock is likely to continue to fluctuate significantly in response to numerous factors, some of which are beyond our control, such as:
| ● | sales of Elfabrio by Chiesi; |
| ● | our sale of shares of our common stock under our ATM program, or market expectations that such sales are to be executed; |
| ● | purchases of BioManguinhos alfataliglicerase in Brazil; |
| ● | the progress and results of the studies of our product candidates under development; |
| ● | the announcement of new products or product enhancements by us or our competitors; |
| ● | developments concerning intellectual property rights and regulatory approvals; |
| ● | variations in our and our competitors’ results of operations; |
| ● | changes in earnings estimates or recommendations by securities analysts; |
| ● | developments in the biotechnology industry; and |
| ● | general market conditions and other factors, including factors unrelated to our operating performance, such as the Israel-Hamas war. |
Continued market fluctuations could result in extreme volatility in the price of our common stock, which could cause a decline in the value of our common stock. Price volatility of our common stock may be worse when the trading volume of our common stock is low. We have not paid, and do not expect to pay, any cash dividends on our common stock as any earnings generated from future operations will be used to finance our operations. As a result, investors will not realize any income from an investment in our common stock until and unless their shares are sold at a profit.
Future sales of our common stock could reduce our stock price.
If our stockholders sell substantial amounts of our common stock, including shares of our common stock underlying our outstanding convertible notes and warrants, or if we sell a substantial amount of our common stock under our ATM program, the market price of our common stock could decrease significantly. The perception in the public market that our existing stockholders might sell shares of common stock may also depress the trading price of our common stock.
A substantial majority of our outstanding shares of our common stock are freely tradable without restriction or further registration under the federal securities laws. In addition, we may sell additional shares of our common stock in the future to raise capital. A substantial number of shares of our common stock are reserved for issuance upon the exercise of stock options, upon conversion of our outstanding convertible notes and upon the exercise of our outstanding warrants. At December 31, 2023, there were outstanding options to purchase common stock issued covering approximately 7.0 million shares of our common stock with a weighted average exercise price of $2.13 per share. Also at December 31, 2023, there were 633,409 shares of common stock available for future for issuance in connection with future grants of incentives under our Amended and Restated Protalix BioTherapeutics, Inc. 2006 Stock Incentive Plan, as amended, approximately 15.2 million shares of common stock reserved for issuance upon conversion of our outstanding 2024 Notes and approximately 13.4 million shares of common stock reserved for issuance upon the exercise of our outstanding warrants. The issuance and sale of substantial amounts of common stock, or the perception that such issuances and sales may occur, could adversely affect the market price of our common stock and impair our ability to raise capital through the sale of additional equity securities.
58
If securities analysts stop publishing research or reports about us or our business or if they downgrade our common stock, the market price of our common stock could decline.
The market for our common stock relies in part on the research and reports that industry or financial analysts publish about us or our business. We do not control these analysts. If any analyst who covers us downgrades our stock or lowers its future stock price targets or estimates of our operating results, the market price for our common stock could decline rapidly. Furthermore, if any analyst ceases to cover us, we could lose visibility in the market, which in turn could cause the market price of our common stock to decline.
Compliance with changing regulation of corporate governance and public disclosure may result in additional expenses, divert management’s attention from operating our business which could have a material adverse effect on our business.
The laws, rules, regulations and standards including the rules promulgated by the national securities exchanges, including the NYSE American, to which we are subject are changed and/or amended from time to time. New or changed laws, rules, regulations and standards are subject to varying interpretations in many cases due to their lack of specificity, and as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies, which could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. As a result, our efforts to comply with evolving laws, rules, regulations and standards are likely to continue to result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. Members of our Board of Directors and our executive officers, could face an increased risk of personal liability in connection with the performance of their duties. As a result, we may have difficulty attracting and retaining qualified directors and executive officers, which could have a material adverse effect on our business. If our efforts to comply with new or changed laws, regulations and standards differ from the activities intended by regulatory or governing bodies, we may incur additional expenses to comply with standards set by regulatory authorities or governing bodies which may have a material adverse effect on our business, results of operations and financial condition.
The issuance of preferred stock or additional shares of common stock could adversely affect the rights of our stockholders.
Our Board of Directors is authorized to issue up to 100,000,000 shares of preferred stock without any further action on the part of our stockholders. Our Board of Directors has the authority to fix and determine the voting rights, rights of redemption and other rights and preferences of preferred stock. Currently, we have no shares of preferred stock outstanding.
Our Board of Directors may, at any time, authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of the shares, together with a premium, before the redemption of our common stock, which may have a material adverse effect on the rights of the holders of our common stock. In addition, our Board of Directors, without further stockholder approval, may, at any time, issue large blocks of preferred stock. In addition, the ability of our Board of Directors to issue shares of preferred stock without any further action on the part of our stockholders may impede a takeover of our company and may prevent a transaction that is favorable to our stockholders.
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
Risk Management and Strategy
Our operations include the creation, collection and maintenance of sensitive information, including proprietary and confidential business information, intellectual property, third-party information and employee information. To protect this
59
information, we use managed detection and response services to monitor our network infrastructure and associated endpoints for possible cybersecurity threats on a constant basis. In addition, we use multi-factor authentication (MFA) for external use, perform penetration testing and engage third parties to assess the effectiveness of our cybersecurity practices. We conduct a thorough risk assessment by identifying critical assets, recognizing potential threats and vulnerabilities, and implement strategies to mitigate these risks and their possible impacts. We establish incident response plans and provide cybersecurity training to our employees and monitor their activity to ensure adherence to our security protocols. A material cyber-attack on our systems, or any other third-party partners or vendors and their key operating systems, may interrupt our ability to operate our business, damage our reputation, or result in monetary damages.
We have implemented a Data Protection Policy, or the DPP, in order to establish the high-level direction for properly managing the use, privacy, security, retention, and disposal of our information, data and assets, and to manage identified material cybersecurity risks. The DPP was prepared using relevant guidance issued and technology standards that are used across various industries. It applies to all entities who are using our equipment and resources, including but not limited to, employees and temporary workers. Our Senior Director, Information Technology, is primarily responsible for implementing and overseeing the DPP and identifying, measuring, monitoring, and reporting on key enterprise-wide risks, including cybersecurity risks. He is expected to become a certified Information Security Officer in March 2024.
Our DPP includes an incident response process that includes reporting thresholds and follows standardized identification and authentication practices. If an incident is identified, it is documented by our Senior Director, Information Technology, who is required to report the incident to management.
We work with third-party service providers from time to time that assist us to identify, assess and manage cybersecurity risks, including professional SEIM SOC and other services firms, threat intelligence service providers, cybersecurity consultants, cybersecurity software providers, managed cybersecurity service providers, and penetration testing.
No risks from cybersecurity threats have occurred that have affected our business, results of operations or financial condition.
Governance
Our cybersecurity risk assessment and management processes are implemented and maintained by certain members of our management, including our Senior Director, Information Technology, who reports to our Sr. Vice President, Operations. Management is also responsible for hiring appropriate personnel, integrating cybersecurity considerations into our overall risk management strategy, and for communicating key priorities to employees, as well as for approving budgets, helping prepare for cybersecurity incidents, approving cybersecurity processes, and reviewing security assessments and other security-related reports. Our incident response process involves management, who participates in our disclosure controls and procedures.
Our incident response process is designed to escalate certain cybersecurity incidents and vulnerabilities to members of management depending on the circumstances, including work with our incident response team to help the company mitigate and remediate cybersecurity incidents of which they are notified. In addition, the company’s incident response processes include reporting to our Board of Directors for certain cybersecurity incidents.
Management is involved with our efforts to prevent, detect, and mitigate cybersecurity incidents by overseeing preparation of cybersecurity policies and procedures, testing incident response plans and engaging vendors to conduct penetration tests. Management participates in cybersecurity incident response efforts by being a member of the incident response team and helping direct our response to cybersecurity incidents.
Our Board of Directors addresses our cybersecurity risk management as part of its general oversight function. Our Chief Financial Officer and IT consultant provide periodic briefings to the Board of Directors regarding our cybersecurity risks and activities, including any recent cybersecurity incidents and related responses, if any, cybersecurity systems testing, activities of third parties, and the like.
60
security, nondisclosure of confidential information or disclosure of sensitive data or fail or are perceived to fail to comply with applicable data protection laws and regulations, or consumer protection laws, regulations and standards.”
Item 2. Properties
We maintain a U.S. corporate office in Hackensack, New Jersey. Our headquarters, including a manufacturing facility, executive offices and other facilities, are located in Carmiel, Israel. Our facilities in Israel currently contain approximately 14,700 sq/ft of manufacturing space and 3,400 sq/ft for a pilot plant, 11,700 sq/ft for offsite warehouse space and approximately 43,100 sq/ft of laboratories, front warehouse and office space, and are leased at a rate of approximately $83,000 per month. In addition, we are entitled to use an additional 14,500 sq/ft in the same facility, which we intend to utilize in connection with an anticipated expansion of our manufacturing facilities. Our facilities are equipped with the requisite laboratory services required to conduct our business, and we believe that the existing facilities are adequate to meet our needs for the foreseeable future. Our original lease for the facility was in effect until 2016 and we have exercised two of three options to extend the term, each for an additional five-year period. The lease is currently in effect until 2026 and we retain an additional option to extend the term for another five-year period thereafter. Upon the exercise of each option to extend the term of the lease includes a 10% increase to the then current base rent.
Item 3. Legal Proceedings
We are not involved in any material legal proceedings.
Item 4. Mine Safety Disclosures
Not applicable.
61
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is traded on the NYSE American under the symbol “PLX.” In 2023, we decided to voluntarily delist our common stock from the Tel Aviv Stock Exchange.
Holders
As of March 1, 2024, there were approximately 61 holders of record of our common stock. A substantially greater number of holders of our common stock are “street name” or beneficial holders, whose shares are held of record by banks, brokers and other financial institutions.
Dividend Policy
To date, we have not declared or paid any cash dividends on our common stock. We do not anticipate paying any dividends on our common stock in the foreseeable future.
Securities Authorized for Issuance under Equity Compensation Plans
The following table provides information as of December 31, 2023 with respect to the shares of our common stock that may be issued under our existing equity compensation plan.
| A |
| B |
| C | ||
Number of Securities Remaining | |||||||
Number of Securities | Available for Future Issuance | ||||||
to be Issued | Weighted Average | Under Equity Compensation Plans | |||||
Upon Exercise of | Exercise Price of | (Excluding Securities Reflected in | |||||
Plan Category | Outstanding Options | Outstanding Options | Column A) | ||||
Equity Compensation Plans Approved by Stockholders |
| 6,965,601 | $ | 2.13 |
| 633,409 | |
Equity Compensation Plans Not Approved by Stockholders |
| — |
| — |
| — | |
Total |
| 6,965,601 | $ | 2.13 |
| 633,409 | |
Recent Sales of Unregistered Securities
None.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Item 6. [Reserved]
62
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations (MD&A) is intended to help the reader understand our results of operations and financial condition. The MD&A is provided as a supplement to, and should be together with our consolidated financial statements and the related notes included elsewhere in this Annual Report on Form 10-K. Some of the information contained in this discussion and analysis, particularly with respect to our plans and strategy for our business and related financing, include forward-looking statements that involve risks and uncertainties. You should read “Risk Factors” in Item 1A of this Annual Report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a biopharmaceutical company focused on the development and commercialization of recombinant therapeutic proteins primarily based on our proprietary ProCellEx protein expression system. To date, we have successfully developed two ERTs; Elfabrio (pegunigalsidase alfa) for the treatment of adult patients with a confirmed diagnosis of Fabry disease and Elelyso (taliglucerase alfa) for the treatment of adult patients with Gaucher disease. Elfabrio, which we referred to as PRX-102 during its development stage, has been approved for marketing in the United States, the European Union, Great Britain, Switzerland and Israel. Chiesi is our commercialization partner for Elfabrio.
We have licensed the rights to commercialize Elelyso worldwide (other than Brazil) to Pfizer, and in Brazil to Fiocruz, an arm of the Brazilian MoH. Elelyso is marketed as BioManguinhos alfataliglicerase in Brazil.
Our strategy is to develop proprietary recombinant proteins designed to address high, unmet needs in the rare disease space that are therapeutically superior to existing recombinant proteins currently marketed for the same indications. Consistent with this strategy, we have a number of product candidates in varying stages of the clinical development process.
On May 5, 2023, the EC announced that it had approved the MAA for Elfabrio and on May 9, 2023, the FDA announced that it had approved the BLA for Elfabrio, each for adult patients with a confirmed diagnosis of Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. The EMA approval followed the February 2023 adoption of a positive opinion and recommendation of marketing authorization for Elfabrio by the CHMP. Elfabrio was approved by the FDA with a boxed warning for hypersensitivity reactions/anaphylaxis, consistent with Enzyme Replacement Therapy (ERT) class labeling, and Warnings/Precautions providing guidance on the signs and symptoms of hypersensitivity and infusion-associated reactions seen in the clinical studies as well as treatments to manage such events should they occur. The Warnings/Precautions for membranoproliferative glomerulonephritis (MPGN) alert prescribers to the possibility of MPGN and provide guidance for appropriate patient management. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease.
We have entered into two exclusive global licensing and supply agreements for Elfabrio with Chiesi. In October 2017 and July 2018, Protalix Ltd., our wholly-owned subsidiary, entered into the Chiesi Agreements pursuant to which Chiesi was granted an exclusive license for all markets, both within and outside of the United States to commercialize Elfabrio.
Elfabrio was the subject of a phase III clinical program studying the drug as a treatment of patients with Fabry disease, a rare, genetic lysosomal disorder. The phase III clinical program included three separate studies, the BALANCE study, the BRIDGE study and the BRIGHT study. The phase III clinical program analyzed two potential dosing regimens: 1 mg/kg every two weeks and 2 mg/kg every four weeks. In addition, the phase III clinical program included two extension studies in which subjects that participated in our phase I/II clinical trials and phase III clinical trials had the opportunity to enroll and continue to be treated with PRX-102. As of March 1, 2023, sponsorship of the two open label extension studies was transferred to Chiesi, which is now administering the extension studies. From time to time, and as Elfabrio is approved for marketing in different jurisdictions, participants withdraw from the open label extension studies. Some of the withdrawals transfer to a commercial setting, others withdraw for other reasons.
63
The BLA for Elfabrio for the treatment of adult patients with Fabry disease was resubmitted to the FDA on November 9, 2022. An initial BLA for Elfabrio was submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, but resulted in a CRL.
The MAA was submitted to the EMA on February 7, 2022, after the October 8, 2021 meeting we held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102.
The FDA publicly released the internal review documents for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study with confirmatory evidence provided by the BALANCE study. The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks. However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support the non-inferiority margin.
We continuously evaluate potential strategic marketing partnerships as well as collaboration programs with biotechnology and pharmaceutical companies and academic research institutions. Except with respect to Elfabrio and Elelyso, we hold the worldwide commercialization rights to our other proprietary development candidates.
Our product pipeline currently includes, among other candidates:
Obtaining marketing approval with respect to any product candidate in any country is dependent on our ability to implement the necessary regulatory steps required to obtain such approvals, and demonstrate the safety and efficacy of its product candidates. We cannot reasonably predict the outcome of these activities.
On March 21, 2023, the first patient was dosed in our phase I First-in-Human (FIH) clinical trial of PRX-115. As of December 31, 2023, 56 patients have been dosed in this trial. We expect to publish preliminary results of this study during the second quarter of 2024.
On July 2, 2021, we entered into an At The Market Offering Agreement, or the 2021 Sales Agreement, with H.C. Wainwright & Co., LLC, as our sales agent, or the Agent, which was amended on May 2, 2022. Pursuant to the terms of the 2021 Sales Agreement, we were able to sell from time to time through the Agent shares of our common stock having an aggregate offering price of up to $20.0 million, or the ATM Shares. Upon execution of the 2021 Sales Agreement, we terminated the ATM Equity OfferingSM Sales Agreement it had entered into on October 1, 2020 with BofA Securities, Inc., or BofA Securities. During the term of the sales agreement with BofA Securities, we sold a total of 3,296,123 shares of common stock for total gross proceeds of approximately $13.8 million.
During the term of the 2021 Sales Agreement which ended during the quarter ended March 31, 2023, we sold a total of 13,980,060 ATM Shares for total gross proceeds of approximately $20.0 million, thereby completing the ATM program under said agreement.
On February 27, 2023, we entered into an At The Market Offering Agreement, or the 2023 Sales Agreement, with the Agent. Pursuant to the terms of the 2023 Sales Agreement, we may sell, from time to time through the Agent, ATM Shares having an aggregate offering price of up to $20.0 million. As of December 31, 2023, shares of Common Stock for total gross proceeds of approximately $6.4 million remain available to be sold under the 2023 Sales Agreement.
Under each of the Chiesi Agreements, Chiesi made an upfront payment to Protalix Ltd. of $25.0 million in connection with the execution of each agreement. In addition, under the Chiesi Ex-US Agreement, Protalix Ltd. was entitled to additional payments of up to $25.0 million in pegunigalsidase alfa development costs, and to receive additional
64
payments of up to $320.0 million, in the aggregate, in regulatory and commercial milestone payments. Under the Chiesi US Agreement, Protalix Ltd. is entitled to payments of up to a maximum of $20.0 million to cover development costs for pegunigalsidase alfa, and to receive additional payments of up to a maximum of $760.0 million, in the aggregate, in regulatory and commercial milestone payments. To date, Protalix Ltd. has received the complete amount of development costs to which it is entitled under the Chiesi Agreements. In addition, following the approval of Elfabrio by the FDA, we received a milestone payment equal to $20.0 million.
On May 13, 2021, we signed a binding term sheet with Chiesi pursuant to which we and Chiesi amended the Chiesi Agreements in order to provide us with near-term capital. Chiesi agreed to make a $10.0 million payment to us before the end of the second quarter of 2021 in exchange for a $25.0 million reduction in a longer term regulatory milestone payment provided in the Chiesi Ex-US Agreement. All other regulatory and commercial milestone payments remain unchanged. We received the payment in June 2021. We also agreed to negotiate certain manufacturing related matters.
Under the terms of both of the Chiesi Agreements, Protalix Ltd. is required to manufacture all of the Elfabrio drug substance needed under the agreements, subject to certain exceptions, and Chiesi will purchase Elfabrio drug product from Protalix, subject to certain terms and conditions. The consideration for Protalix Ltd. is based on the drug product supplied to Chiesi and the average selling price of the drug product in the relevant territory multiplied by tiered payments as described in the relevant agreement. Under the Chiesi Ex-US Agreement, the price payable to us for drug product supplied is based on a range of 15% to 35% of the average selling price of the drug product in the applicable territory, and, under the Chiesi US Agreement, such price is based on a range of 15% to 40% of the average selling price of the drug product in the United States.
On August 29, 2022, we entered into a Fill/Finish Agreement, or the F/F Agreement, and a Letter Agreement, or the Letter Agreement, in each case with Chiesi. We agreed to supply Chiesi with drug substance for Elfabrio and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide us with commercial fill/finish services for Elfabrio, including to support the anticipated global launch of Elfabrio. The F/F Agreement shall continue in force until December 31, 2025, unless terminated earlier in accordance with the terms of the F/F Agreement and the term may be extended by mutual agreement for an additional period of seven years upon mutual written agreement prior to expiration of the initial term.
Since its approval by the FDA, Elelyso has been marketed by Pfizer in accordance with the Pfizer Agreement. In October 2015, Protalix Ltd. and Pfizer entered into the Amended Pfizer Agreement, pursuant to which we sold to Pfizer our share in the collaboration created under the Pfizer Agreement for the commercialization of Elelyso. As part of the sale, we agreed to transfer our rights to Elelyso in Israel to Pfizer while gaining full rights to it in Brazil. Under the Amended Pfizer Agreement, Pfizer is entitled to all of the revenues, and is responsible for 100% of expenses globally for Elelyso, excluding Brazil where we are responsible for all expenses and retain all revenues.
On June 18, 2013, we entered into the Brazil Agreement. Fiocruz’s purchases of BioManguinhos alfataliglicerase to date have been significantly below certain agreed-upon purchase milestones and, accordingly, we have the right to terminate the Brazil Agreement. Notwithstanding the termination right, we are, at this time, continuing to supply BioManguinhos alfataliglicerase to Fiocruz and patients continue to be treated with BioManguinhos alfataliglicerase in Brazil.
Because our operations are conducted in the State of Israel, the business and operations may be directly affected by economic, political, geopolitical and military conditions in Israel. In October 2023, Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched extensive rocket attacks on Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel attacking a number of civilian and military targets while simultaneously launching extensive rocket attacks on the Israeli population and industrial centers. At the same time, clashes between Israel and Hezbollah in Lebanon have increased. In response, Israel’s security cabinet declared war against the Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks. Moreover, the attacks by Hamas and Hezbollah, and Israel’s defensive measures, may result in a greater regional conflict. It is currently not possible to predict the duration or severity of the ongoing conflict or its effects on our business, operations and financial conditions. The ongoing conflict is rapidly evolving and developing, and could disrupt certain of our business and operations, among others. As of the issuance of these financial statements, the impact of the war has not had a material adverse effect on our operations.
65
We believe that our cash and cash equivalents and short-term bank deposits as of December 31, 2023 are sufficient to satisfy our capital needs for at least 12 months from the date that these financial statements are issued.
Critical Accounting Policies
Our significant accounting policies are more fully described in note 1 to our consolidated financial statements appearing at the end of this Annual Report on Form 10-K. We believe that the accounting policies below are critical for one to fully understand and evaluate our financial condition and results of operations.
The discussion and analysis of our financial condition and results of operations is based on our financial statements, which we prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate such estimates and judgments, including those described in greater detail below. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Revenues
Our primary sources of revenues include our sales of BioManguinhos alfataliglicerase in Brazil, of drug substance to Pfizer under our Amended Pfizer Agreement and of drug product to Chiesi under the Chiesi Agreements. We recognize revenue from selling goods at a point in time when control over the product is transferred to customers (upon delivery), at the net selling price, which reflects sales reserves for variable consideration, potential discounts, rebates and allowances. The sales reserve is calculated based on expected sales and the anticipated net average selling price.
The transaction price is the consideration to which we expect to be entitled from the customer. The consideration promised in a contract with our customers may include fixed amounts and variable amounts. We estimate the variable consideration and includes it in the transaction price using the most likely outcome method, and only to the extent the variable consideration is probable that a significant reversal of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Prior to recognizing revenue from variable consideration, we use significant judgment to determine the probability of significant reversal of such revenue.
We also generate revenues from the Chiesi Agreements. According to Accounting Standards Codification 606, Revenue from Contracts with Customers, and all the related amendments, or ASC 606, a performance obligation is a promise to provide a distinct good or service or a series of distinct goods or services. Goods and services that are not distinct are bundled with other goods or services in the contract until a bundle of goods or services that is distinct is created. A good or service promised to a customer is distinct if the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer and the entity’s promise to transfer the good or service to the customer is separately identifiable from other promises in the contract.
We have identified two performance obligations in the Chiesi Agreements as follows: (1) the license and research and development services and (2) a contingent performance obligation regarding future manufacturing.
We determined that the license together with the research and development services should be combined into a single performance obligation since Chiesi cannot benefit from the license without the research and development services. The research and development services are highly specialized and are dependent on the supply of the drug.
The future manufacturing was contingent on regulatory approvals of the drug and we deem these services to be separately identifiable from other performance obligations in the contract. Manufacturing services post-regulatory approval are not interdependent or interrelated with the license and research and development services. Following the regulatory approvals for Elfabrio received in May 2023, we started recognizing revenue from manufacturing.
The transaction price of the Chiesi Agreements was comprised of fixed consideration and variable consideration (capped research and development reimbursements). Under ASC 606, the consideration to which we would be entitled upon the
66
achievement of contractual milestones, which are contingent upon the occurrence of future events, are a form of variable consideration. We estimate variable consideration using the most likely method. Amounts included in the transaction price are recognized only when it is probable that a significant reversal of cumulative revenues will not occur. Prior to recognizing revenue from variable consideration, we use significant judgment to determine the probability of a significant reversal of such revenue. Following the approval of Elfabrio by the FDA, we received a milestone payment equal to $20.0 million.
Since the customer benefits from the research and development services as the entity performs the service, revenue from granting the license and the research and development services is recognized over time using the cost-to-cost method.
Revenue from additional research and development services ordered by Chiesi is recognized over time using the cost-to-cost method.
Income Taxes
We estimate the degree to which deferred tax assets will result in a benefit, after consideration of all positive and negative evidence, and provide a valuation allowance for deferred tax assets that we believe more likely than not will not be realized. In situations in which we are able to determine that our deferred tax assets will be realized, that determination generally relies on future reversals of taxable temporary differences and expected future taxable income. Significant judgment is required in determining any valuation allowance recorded against deferred tax assets. In assessing the need for a valuation allowance, we considered all available evidence, including past operating results, the most recent projections for taxable income, and prudent and feasible tax planning strategies. We reassess our valuation allowance periodically and if future evidence allows for a partial or full release of the valuation allowance, we reverse the related valuation allowance. The tax valuation allowance totaled approximately $56.0 million at December 31, 2023 (see Note 13 to our consolidated financial statements for additional information). Should our actual future taxable income by tax jurisdiction vary from estimates, additional allowances or reversals thereof may be necessary.
Significant judgment is required in evaluating our uncertain tax positions. In evaluating the exposure associated with our various tax filing positions, we record reserves for uncertain tax positions in accordance with U.S. GAAP based on the technical support for the positions and our past audit experience with similar positions. For those tax positions where it is more likely than not that a tax benefit will be sustained, we record the largest amount of tax benefit with a greater than 50% likelihood of being realized upon ultimate settlement with a taxing authority that has full knowledge of all relevant information. We believe our tax positions comply with applicable tax laws and we intend to defend our positions, no assurance can be given that the final tax outcome of these matters will not be different from that which is reflected in our historical income tax reserves and accruals. To the extent that the final tax outcome of these matters is different than the amounts recorded, such differences will impact the provision for income taxes in the period in which such determination is made. The provision for income taxes includes the impact of reserve provisions and changes to reserves that are considered appropriate. Our liability for these unrecognized tax benefits totaled approximately $0.8 million at December 31, 2023 (see Note 13 to our consolidated financial statements for additional information).
Research and Development Expense
We expect our research and development expense to remain our primary expense in the near future as we continue to develop our product candidates. Research and development expense consists of:
| ● | internal costs associated with research and development activities; |
| ● | payments made to third party contract research organizations, investigative/clinical sites and consultants; |
| ● | manufacturing development costs; |
| ● | personnel-related expenses, including salaries, benefits, travel and related costs for the personnel involved in research and development; |
| ● | activities relating to the advancement of product candidates through preclinical studies and clinical trials; and |
67
| ● | facilities and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities, as well as laboratory and other supplies. |
The following table identifies our current major research and development projects:
Project |
| Status |
| Expected Near Term Milestones |
PRX-115 – PEGylated Uricase |
| Phase I |
| Preliminary results from the phase I clinical trial expected in the second quarter of 2024. |
|
|
|
| |
PRX-119 – Long Acting DNase I |
| Preclinical |
|
We anticipate incurring increasing costs in connection with the continued development of PRX-115 and PRX-119 and in the expansion of our pipeline. Our internal resources, employees and infrastructure are not tied to any individual research project and are typically deployed across all of our projects. We currently do not record and maintain research and development costs per project.
At this time, due to the inherently unpredictable nature of preclinical and clinical development processes and given the early stage of our preclinical product development programs, we are unable to estimate with any certainty the costs we will incur in the continued development of the product candidates in our pipeline for potential commercialization. Clinical development timelines, the probability of success and development costs can differ materially from expectations. Our future research and development expenses for our product candidates will depend on the preclinical and clinical success of each product candidate, as well as ongoing assessments of each product candidate’s commercial potential. In addition, we cannot forecast with any degree of certainty which product candidates may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements. See “Risk Factors—We may not obtain the necessary U.S., EMA or other worldwide regulatory approvals to commercialize our drug candidates in a timely manner, if at all, which would have a material adverse effect on our business, results of operations and financial condition.”
We expect our research and development expenses to continue to be our primary expense in the future as we continue the advancement of our clinical trials and preclinical product development programs for our product candidates. The lengthy process of completing clinical trials and seeking regulatory approvals for our product candidates requires expenditure of substantial resources. Any failure or delay in completing clinical trials, or in obtaining regulatory approvals, could cause a delay in generating product revenue and cause our research and development expense to increase and, in turn, have a material adverse effect on our operations. Due to the factors set forth above, we are not able to estimate with any certainty when we would recognize any net cash inflows from our projects. See “Risk Factors—Clinical trials are very expensive, time-consuming and difficult to design and implement and may result in unforeseen costs, which may have a material adverse effect on our business, results of operations and financial condition.”
Share-Based Compensation
We measure share-based compensation cost for all share-based awards at the fair value on the grant date and recognition of share-based compensation over the related service period. The fair value of stock options is determined based on the number of shares granted and the price of our common stock, and calculated based on the Black-Scholes valuation model. For grants made to employees and non-employees, we recognize the fair value of the grant as expense over the service period using the accelerated method.
The guidance requires companies to estimate the expected term of the option rather than simply using the contractual term of an option. Because of lack of sufficient data on past option exercises by employees, the expected term of the options could not be based on historic exercise patterns. Accordingly, we adopted the simplified method, according to which companies may calculate the expected term as the average between the vesting date and the expiration date, assuming the option was granted as a “plain vanilla” option.
In performing the valuation, we assumed an expected 0% dividend yield in the previous years and in the next years. We do not have a dividend policy and given the lack of profitability, dividends are not expected in the foreseeable future, if
68
at all. The guidance stipulates a number of factors that should be considered when estimating the expected volatility, including the implied volatility of traded options, historical volatility and the period that the shares of the company are being publicly traded.
The risk-free interest rate used in the valuation of the options is based on the implied yield of U.S. federal reserve zero–coupon government bonds. The remaining term of the bonds used for each valuation was equal to the expected term of the grant. This methodology has been applied to all grants valued by us. The guidance requires the use of a risk–free interest rate based on the implied yield currently available on zero–coupon government issues of the country in whose currency the exercise price is expressed, with a remaining term equal to the expected life of the option being valued. This requirement has been applied for all grants valued as part of this report.
Convertible Notes
The outstanding convertible notes are accounted for using the guidance set forth in the Financial Accounting Standards Board, or the FASB, Accounting Standards Codification, or ASC, 815 requiring that we determine whether the embedded conversion option must be separated and accounted for separately. ASC 470-20 regarding debt with conversion and other options requires the issuer of a convertible debt instrument that may be settled in cash upon conversion to separately account for the liability (debt) and equity (conversion option) components of the instrument in a manner that reflects the issuer’s nonconvertible debt borrowing rate. Our Senior Secured 7.50% Convertible Notes Due 2021, or the 2021 Notes, were accounted for partially as liability and equity components of the instrument and partially as a debt host contract with an embedded derivative resulting from the conversion feature. The 2024 Notes were accounted for as a liability (debt) and equity component (conversion option) as the convertible notes may be settled wholly or partly in cash, at our option, when converted.
Issuance costs regarding the issuance of the 2021 Notes, as well as the debt discount and debt issuance costs from the issuance of the 2024 Notes, were deferred and amortized over the applicable convertible notes period using the effective interest rate.
On August 25, 2021, we completed the exchanges of a substantial majority of the 2021 Notes with certain institutional note holders, or the Exchanges. The Exchanges involved the exchange of an aggregate of $54.65 million principal amount of our outstanding 2021 Notes for an aggregate of $28.75 million principal amount of newly issued 2024 Notes, $25.90 million in cash, and approximately $1.1 million in cash representing accrued and unpaid interest through the closing date. The initial conversion rate for the 2024 Notes is 563.2216 shares of common stock for each $1,000 principal amount of 2024 Notes (equivalent to an initial conversion price of approximately $1.7755 per share of the common stock), subject to adjustment in certain circumstances. This initial conversion price represents a premium of approximately 32.5% relative to the closing price of our common stock on the NYSE American on August 13, 2021. The Exchanges are described in greater detail in Note 10 to the consolidated financial statements.
For accounting purposes, as the terms of the 2021 Notes and the 2024 Notes are substantially different, the Exchanges were considered an extinguishment of debt. We allocated the fair value of the consideration transferred to the participating note holders between the 2021 Notes and their equity component based on the fair value of the liability component before the extinguishment, and the remainder was allocated to the equity component. As a result, we recognized a loss from extinguishment in the statement of operations equal to $0.8 million due to derecognition of the liability component and a reduction of stockholders’ equity of $12.2 million.
As of December 31, 2023, a total of $20.4 million aggregate principal amount of the 2024 Notes were outstanding. In addition, as of December 31, 2023, 2022 and 2021, none of the 2021 Notes were outstanding.
Results of Operations
Year ended December 31, 2023 Compared to the Year Ended December 31, 2022
Revenues from Selling Goods
We recorded revenues from selling goods of $40.4 million for the year ended December 31, 2023, an increase of $15.1 million, or 60%, compared to revenues of $25.3 million for the year ended December 31, 2022. The increase
69
resulted primarily from an increase of $14.1 million in sales of Elfabrio drug product to Chiesi, following the approvals by the FDA and the EMA of Elfabrio, an increase of $0.1 million in sales to Pfizer and of $0.9 million in sales to Brazil, resulting from timing differences.
Revenues from License and R&D services
We recorded revenues from license and R&D services of $25.1 million for the year ended December 31, 2023, an increase of $2.8 million, or 13%, compared to revenues of $22.3 million for the year ended December 31, 2022. The increase resulted from the $20.0 million regulatory milestone payment from Chiesi in connection with the FDA approval of Elfabrio which was partially offset by a decrease of $17.2 million in revenues recognized in connection with the R&D performance obligation under the Chiesi Agreements as we have completed the phase III clinical program thereunder. Revenues from license and R&D services represent mainly the revenues we recognized in connection with the Chiesi Agreements.
Cost of Goods Sold
Cost of goods sold was $23.0 million for the year ended December 31, 2023, an increase of $3.4 million, or 17%, compared to cost of goods sold of $19.6 million for the year ended December 31, 2022. The increase in cost of goods sold was primarily the result of the increase in sales of goods to Chiesi, Brazil and to Pfizer. Sales to Chiesi included certain drug substance costs which had already been recognized as research and development expenses as it was produced as part of research and development activities. Accordingly, the related cost of goods sold does not include the costs of such drug substance.
Research and Development Expenses
For the year ended December 31, 2023, our total research and development expenses were approximately $17.1 million comprised of approximately $6.3 million in subcontractor-related expenses, approximately $7.8 million of salary and related expenses, approximately $0.6 million of materials-related expenses and approximately $2.4 million of other expenses. For the year ended December 31, 2022, our total research and development expenses were approximately $29.3 million comprised of approximately $17.8 million in subcontractor-related expenses, approximately $7.3 million of salary and related expenses, approximately $1.4 million of materials-related expenses and approximately $2.8 million of other expenses.
Total decrease in research and developments expenses was $12.2 million, or 42%, for the year ended December 31, 2023 compared to the year ended December 31, 2022. The decrease in research and development expenses resulted primarily from a $11.5 million decrease in subcontractor-related expenses in connection with our PRX-102 clinical trials, and a $0.8 million decrease in materials-related expenses.
We expect research and development expenses to continue to be our primary expense as we enter into a more advanced stage of preclinical and clinical trials for certain of our product candidates.
Selling, General and Administrative Expenses
Selling, general and administrative expenses were $15.0 million for the year ended December 31, 2023, an increase of $3.3 million, or 28%, from $11.7 million for the year ended December 31, 2022. The increase resulted primarily from an increase of approximately $2.3 million in one-time cash bonuses, share-based compensation and salary and salary-related expenses, as well as an increase of $0.3 million in travel, conferences and employee training expenses.
Financial Expenses and Income, Net
Financial expense, net was $1.9 million for the year ended December 31, 2023, an increase of $0.5 million, or 36%, compared to financial expenses of $1.4 million for the year ended December 31, 2022. The increase was primarily due to a decrease of $0.9 million in income related to exchange rates as well as an increase in our interest expenses of $0.7 million which was partially offset by a gain recognized due to conversions of a portion of the 2024 Notes of $0.4 million and a $0.6 million increase in interest income.
70
Income taxes
For the year ended December 31, 2023, we recorded income taxes of approximately $0.3 million, a decrease of $0.2 million, or 40%, compared to tax expenses of $0.5 million for the year ended December 31, 2022. The income taxes resulted primarily from the provision for current taxes on income mainly derived from GILTI income mainly in respect of Section 174 of the TCJA. Effective in 2022, Section 174 of the TCJA requires all U.S. companies, for tax purposes, to capitalize and subsequently amortize R&D expenses that fall within the scope of Section 174 over five years for research activities conducted in the United States and over 15 years for research activities conducted outside of the United States rather than deducting such costs in the current year. The net income taxes gives effect to a valuation allowance release equal to approximately $3.1 million.
Year ended December 31, 2022 Compared to the Year Ended December 31, 2021
For a discussion of the year ended December 31, 2022 compared to the year ended December 31, 2021, see Management’s Discussion and Analysis of Financial Condition and Results of Operations included in our Annual Report on Form 10-K for the year ended December 31, 2022.
Liquidity and Capital Resources
Our sources of liquidity include our cash balances and bank deposits. At December 31, 2023, we had $44.6 million in cash and cash equivalents and short term bank deposits. We have primarily financed our operations through equity and debt financings, business collaborations, and grants funding.
During the year ended December 31, 2023, we raised gross proceeds equal to approximately $24.9 million from the sale of 12,560,150 shares of our common stock under our ATM program. All such sales were completed during the quarters ended March 31, 2023 and June 30, 2023. No sales were completed during the quarters ended September 30, 2023 and December 31, 2023.
During the year ended December 31, 2021, we raised gross proceeds equal to approximately $8.8 million from sales of common stock under our ATM program through the sale of 1,867,552 shares of our common stock. In addition, we raised gross proceeds of approximately $40.2 million from a public offering of our common stock before deducting the underwriting discount and estimated expenses of the offering. In connection with the offering, we issued 8,749,999 shares of our common stock at a purchase price per share of $4.60.
On August 25, 2021, we completed Exchanges with institutional note holders of a substantial majority of the then outstanding 2021 Notes. The Exchanges involved the exchange of an aggregate of $54.65 million principal amount of 2021 Notes for an aggregate of $28.75 million principal amount of newly issued 2024 Notes, $25.90 million in cash and approximately $1.1 million in cash representing accrued and unpaid interest through the closing date. The initial conversion rate of the 2024 Notes is 563.2216 shares of our common stock per $1,000 principal amount of 2024 Notes, which is equivalent to an initial conversion price of approximately $1.7755 per share of common stock, subject to adjustment in certain circumstances. This initial conversion price represents a premium of approximately 32.5% relative to the closing price of the common stock on the NYSE American on August 13, 2021. After giving effect to the Exchanges, $3.27 million aggregate principal amount of the 2021 Notes remained outstanding. On November 15, 2021, all of the then outstanding 2021 Notes matured and were paid in full.
The 2024 Notes were issued pursuant to the 2024 Indenture which was entered into between us, the guarantors party thereto, The Bank of New York Mellon Trust Company, N.A., as trustee and Wilmington Savings Fund Society, FSB, as collateral agent. Interest on the Notes are payable semi-annually at a rate of 7.50% per annum. The 2024 Notes will mature three years after the issuance thereof, unless earlier purchased, converted, exchanged or redeemed and will be guaranteed by our subsidiaries. The 2024 Notes are secured by perfected liens on all of our assets, including those of our subsidiaries.
Cash Flows
Net cash used in operations was $1.3 million for the year ended December 31, 2023. The net income for the year ended December 31, 2023 of $8.3 million was decreased by a $13.2 million decrease in contracts liability, a $3.1 million
71
increase in deferred tax assets, a $2.2 million increase in inventories, a $0.4 million increase in accounts receivable-trade and other assets and a $0.4 million financial income-net. The net income was increased by a $5.3 million increase in accounts payable and accruals, $3.4 million in share-based compensation and $1.2 million in depreciation. Net cash used in investing activities for the year ended December 31, 2023 was $16.7 million and consisted primarily of net investment in bank deposits. Net cash provided by financing activities was $24.7 million resulting primarily from the sale of common stock under our ATM program.
Future Funding Requirements
Since our inception, we have incurred significant research and development expenditures which have not been offset by revenues. We have not generated significant revenues from sales of Elelyso, and commercial sales of Elfabrio only commenced in the middle of 2023. We have generated operating losses from our continuing operations since our inception although the revenues generated in the year ended December 31, 2023 exceeded our expenditures for the same period.
Under the terms of the 2024 Indenture, we are required to comply with certain covenants, including the requirement to maintain a minimum cash balance of at least $7.5 million. Failure to comply with such covenants may result in an event of default under the 2024 Indenture and, accordingly, may result in the acceleration of the payment of the notes or in additional interest payments. As of December 31, 2023, we were in compliance with all covenants.
As we increase our research and developments efforts with respect to our current and future product candidates, we expect to continue to incur significant expenditures. We cannot anticipate the costs or the timing of the occurrence of such costs. Although we expect the revenues generated from the sales of Elfabrio and Elelyso will increase, such revenues may not be sufficient to fund the expenditures. To the extent we need to obtain additional financing in excess of such anticipated revenues, it may be difficult for us to do so given the volatility of the price of our common stock. Our material cash needs for the next 24 months will include, among other expenses, (i) costs of preclinical and clinical trials, (ii) employee salaries, (iii) payments for rent and operation of our manufacturing facilities, (iv) fees to our consultants and legal advisors, patent advisors and fees for service providers in connection with our research and development efforts, (v) payments of principal and interest on our outstanding 2024 Notes and (vi) tax payments. We believe that the funds currently available to us are sufficient to satisfy our capital needs for at least 12 months.
As discussed above, we may be required to raise additional capital to develop our product candidates and continue research and development activities. Our ability to raise capital, and the amounts of necessary capital, will depend on many other factors, including:
| ● | the duration and cost of discovery and preclinical development and laboratory testing and clinical trials for our product candidates; |
| ● | Chiesi’s progress in commercializing Elfabrio; |
| ● | our progress in commercializing BioManguinhos alfataliglicerase in Brazil; |
| ● | the timing and outcome of regulatory review of our product candidates; |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights; and |
| ● | the costs associated with any litigation claims. |
We expect to finance our future cash needs through sales of Elfabrio and Elelyso, corporate collaborations, licensing or similar arrangements, public or private equity offerings and/or debt financings. We currently do not have any commitments for future external funding, except with respect to the milestone payments that may become payable under the Chiesi Agreements. On February 27, 2023, we entered into the 2023 Sales Agreement pursuant to which we may sell from time to time through the Agent ATM Shares having an aggregate offering price of up to $20.0 million as we had then completed the ATM program under the 2021 Sales Agreement. As of December 31, 2023, shares of our common
72
stock for total gross proceeds of approximately $6.4 million remain available to be sold under the 2023 Sales Agreement. During the quarters ended March 31, 2023 and June 30, 2023, we sold, in the aggregate, 12,560,150 shares of Common Stock under the 2021 Sales Agreement and the 2023 Sales Agreement, generating aggregate gross proceeds equal to approximately $24.9 million.
Contractual obligations
Our contractual obligations include obligations under our convertible notes, operating lease obligations, purchase obligations, certain clinical contracts and the liability for employee rights upon retirement.
Our convertible senior notes had an aggregate outstanding principal balance of $20.42 million at December 31, 2023. The notes will mature on September 1, 2024 unless earlier purchased, converted, exchanged or redeemed, and interest is payable on the note semi-annually at a rate of 7.50% per annum.
We lease certain assets under operating leases, which expire through 2026, with an option to extend the lease on our main facility to 2031. The leases relate primarily to office, laboratory and manufacturing space and vehicles used by our employees. Our aggregate future minimum commitments under these facility and vehicles leases over the next five fiscal years is approximately $4.1 million as of December 31, 2023.
As of December 31, 2023, we are subject to open purchase orders issued to certain suppliers and other vendors mainly in connection with our research and development and manufacturing activities that were outstanding as of December 31, 2023 for approximately $8.1 million over the next five fiscal years.
We have a contractual obligation of approximately $0.4 million as of December 31, 2023 payable over the fiscal year ending December 31, 2024 in connection with contractual arrangements we enter into in the normal course of business with CROs, CMOs and other clinical providers and consultants for clinical trials, preclinical and other research studies and manufacturing services in connection with our primary product development process. These contracts do not contain minimum purchase commitments and are cancelable by us upon prior notice. Payments due upon cancellation consist only of payments for services provided or expenses incurred, including non-cancelable obligations of our service providers, up to the date of cancellation. In addition, we have a $0.1 million commitment payable to a business development consultant.
As of December 31, 2023, we have a contractual obligation of approximately $0.7 million for employee rights upon retirement.
We are also party to certain research and license agreements. If all of the contingencies with respect to milestone payments under our research and license agreements are met, as of December 31, 2023, the aggregate milestone payments payable would be approximately $8.4 million, and would be payable, if at all, as our projects progress over the course of a number of years. The royalty payments payable by our company in connection with sales of each of our product candidates, if any, shall not exceed low, single-digit percentages of net sales of the relevant product.
Effects of Currency Fluctuations
Currency fluctuations could affect us through increased or decreased acquisition costs for certain goods and services. Currency fluctuations during the year ended December 31, 2022, resulted in $1.0 million being recognized as financial income. We do not believe currency fluctuations have had a material effect on our results of operations during the years ended December 31, 2022 or 2023.
Recently Issued Accounting Pronouncements
Certain recently issued and recently adopted accounting pronouncements are discussed in Note 1(r) of the financial statements included in Item 8 of this Annual Report on Form 10-K.
73
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Currency Exchange Risk
The currency of the primary economic environment in which our operations are conducted is the U.S. dollar. Most of our revenues and above 50% of our expenses and capital expenditures are incurred in dollars, and a significant source of our financing has been provided in U.S. dollars. Since the dollar is the functional currency, monetary items maintained in currencies other than the dollar are remeasured using the rate of exchange in effect at the balance sheet dates and non-monetary items are remeasured at historical exchange rates. Revenue and expense items are remeasured at the average rate of exchange in effect during the period in which they occur. Foreign currency translation gains or losses are recognized in the statement of operations.
Approximately 44% of our costs, including salaries, expenses and office expenses, are incurred in NIS. Inflation in Israel may have the effect of increasing the U.S. dollar cost of our operations in Israel. If the U.S. dollar declines in value in relation to the NIS, it will become more expensive for us to fund our operations in Israel. A revaluation of 1% of the NIS will affect our loss before tax by less than 1%. The exchange rate of the U.S. dollar to the NIS, based on exchange rates published by the Bank of Israel, was as follows:
Year Ended December 31, | ||||||||
2021 | 2022 | 2023 | ||||||
Average rate for period | 3.230 | 3.360 | 3.687 | |||||
Rate at period-end | 3.110 | 3.519 | 3.627 | |||||
To date, we have not engaged in hedging transactions. In the future, we may enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rate of the U.S. dollar against the NIS. These measures, however, may not adequately protect us from material adverse effects due to the impact of inflation in Israel.
Interest Rate Risk
Our exposure to market risk is confined to our cash and cash equivalents. We consider all short term, highly liquid investments, which include short-term deposits with original maturities of three months or less from the date of purchase, that are not restricted as to withdrawal or use and are readily convertible to known amounts of cash, to be cash equivalents. The primary objective of our investment activities is to preserve principal while maximizing the interest income we receive from our investments, without increasing risk. We invest any cash balances primarily in bank deposits and investment grade interest-bearing instruments. We are exposed to market risks resulting from changes in interest rates. We do not use derivative financial instruments to limit exposure to interest rate risk. Our interest gains may decline in the future as a result of changes in the financial markets.
Item 8. Financial Statements and Supplementary Data
See the Index to Consolidated Financial Statements on Page F-1 attached hereto.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this Form 10-K. The controls evaluation was conducted under the supervision and with the participation of management, including our Chief Executive Officer and Chief Financial Officer. Disclosure controls and procedures are controls and procedures designed to reasonably assure that information required to be disclosed in our reports filed under the Exchange Act, such as this Form 10-K, is recorded, processed, summarized and
74
reported within the time periods specified in the Commission’s rules and forms. Disclosure controls and procedures are also designed to reasonably assure that such information is accumulated and communicated to our management, including the Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
The evaluation of our disclosure controls and procedures included a review of the controls’ objectives and design, our implementation of the controls and their effect on the information generated for use in this Annual Report on Form 10-K. This type of evaluation will be performed on a quarterly basis so that the conclusions of management, including the Chief Executive Officer and Chief Financial Officer, concerning the effectiveness of the disclosure controls and procedures can be reported in our periodic reports on Form 10-Q and Form 10-K. The overall goals of these various evaluation activities are to monitor our disclosure controls and procedures, and to modify them as necessary. Our intent is to maintain the disclosure controls and procedures as dynamic systems that change as conditions warrant.
Based on the controls evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of the end of the period covered by this Form 10-K, our disclosure controls and procedures were effective to provide reasonable assurance that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified by the Commission, and that material information related to our company and our consolidated subsidiaries are made known to management, including the Chief Executive Officer and Chief Financial Officer, particularly during the period when our periodic reports are being prepared.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles. Internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. generally accepted accounting principles, and that receipts and expenditures of our company are being made only in accordance with authorizations of management and our directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements.
Management assessed our internal control over financial reporting as of December 31, 2023, the end of our fiscal year. Management based its assessment on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Management’s assessment included evaluation of elements such as the design and operating effectiveness of key financial reporting controls, process documentation, accounting policies and our overall control environment.
Based on our assessment, management has concluded that our internal control over financial reporting was effective as of the end of the fiscal year to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with U.S. generally accepted accounting principles. We reviewed the results of management’s assessment with the Audit Committee of our Board of Directors.
Inherent Limitations on Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent or detect all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. The design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Further, because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, within a company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that
75
breakdowns can occur because of simple error or mistake. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Projections of any evaluation of controls effectiveness to future periods are subject to risks. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures.
Attestation Report of Independent Registered Public Accounting Firm
Not applicable.
Changes in internal controls
There were no changes in our internal control over financial reporting (as defined in Rules 13a-15f and 15d-15f under the Exchange Act) that occurred during the quarter ended December 31, 2023 that have materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
Rule 10b5-1 Trading Plansf
During the quarter ended December 31, 2023, none of our directors or officers
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
76
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information in our 2024 Proxy Statement regarding directors and executive officers appearing under the headings “Security Ownership of Certain Beneficial Owners and Management — Section 16(a) Beneficial Ownership Reporting Compliance” and “Election of Directors” is incorporated by reference in this section.
Item 11. Executive Compensation
The information appearing in our 2024 Proxy Statement under the headings “Director Compensation,” “Compensation Discussion and Analysis,” “Report of the Compensation Committee,” and “Executive Compensation” is incorporated by reference in this section.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information appearing in our 2024 Proxy Statement under the heading “Security Ownership of Certain Beneficial Owners and Management” is incorporated by reference in this section.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information appearing in our 2024 Proxy Statement under the headings “Election of Directors—Corporate Governance” and “—Certain Relationships and Related Transactions” is incorporated by reference in this section.
Item 14. Principal Accountant Fees and Services
Our independent registered public accounting firm is Kesselman & Kesselman, Certified Public Accountants (Isr.), a member of PricewaterhouseCoopers International Limited, Tel Aviv, Israel, PCAOB ID. No. 1309.
The information appearing in our 2024 Proxy Statement under the heading “Ratification of Appointment of Independent Registered Public Accounting Firm” is incorporated by reference in this section.
77
PART IV
Item 15. Exhibits and Financial Statement Schedules
The following documents are filed as part of this Annual Report on Form 10-K:
1. Financial Statements. The following Consolidated Financial Statements of Protalix BioTherapeutics, Inc. are included in Item 8 of this Annual Report on Form 10-K:
| Page |
| |
Report of Independent Registered Public Accounting Firm (PCAOB name: Kesselman & Kesselman C.P.A.s and PCAOB ID: 1309) | F-2 | ||
Consolidated Balance Sheets as of December 31, 2022 and 2023 | F-4 | ||
Consolidated Statements of Operations for the years ended December 31, 2021, 2022 and 2023 | F-5 | ||
F-6 | |||
Consolidated Statements of Cash Flows for the years ended December 31, 2021, 2022 and 2023 | F-7 | ||
F-9 | |||
2. Financial Statement Schedule. Financial statement schedules have been omitted since they are either not required, are not applicable or the required information is shown in the consolidated financial statements or related notes.
3. Exhibits.
|
|
|
| Incorporated by Reference |
|
| ||||||
Exhibit |
| Exhibit Description |
| Form |
| File |
| Exhibit |
| Date |
| Filed |
1.1 | 8-K |
| 001-33357 |
| 1.1 |
| February 27, 2023 | |||||
3.1 |
|
| 8-K |
| 001-33357 |
| 3.1 |
| April 1, 2016 |
| ||
|
|
|
|
|
|
| ||||||
3.2 |
|
| Def 14A |
| 001-33357 |
| Appen. A |
| July 1, 2016 |
| ||
|
|
|
|
|
|
| ||||||
3.3 |
| Second Amendment to Certificate of Incorporation of the Company |
| Def 14A |
| 001-33357 |
| Appen. A |
| October 17, 2018 |
| |
|
|
|
|
|
|
| ||||||
3.4 |
| Third Amendment to Certificate of Incorporation of the Company |
| 8-K |
| 001-33357 |
| 3.1 |
| December 20, 2019 |
| |
|
|
|
|
|
|
| ||||||
3.5 |
| Fourth Amendment to Certificate of Incorporation of the Company |
| 10-Q |
| 001-33357 |
| 3.1 |
| August 15, 2022 |
| |
3.6 |
| Fifth Amendment to Certificate of Incorporation of the Company |
| 10-Q |
| 001-33357 |
| 3.6 |
| August 7, 2023 | ||
3.7 |
|
| 8-K |
| 001-33357 |
| 3.2 |
| October 17, 2018 |
| ||
|
|
|
|
|
|
| ||||||
4.1† |
|
| 8-K |
| 001-33357 |
| 4.1 |
| July 18, 2012 |
| ||
78
4.2 | 8-K | 001-33357 |
| 4.1 |
| March 12, 2020 | ||||||
4.3† | 10-Q | 001-33357 |
| 4.8 | August 10, 2020 | |||||||
4.4 | 10-Q | 001-33357 |
| 4.9 | August 10, 2020 | |||||||
4.5 | 8-K |
| 001-33357 |
| 4.2 | August 26, 2021 | ||||||
4.6 | 8-K |
| 001-33357 |
| 4.3 | August 26, 2021 | ||||||
4.7 | X | |||||||||||
10.1 |
|
| 8-K |
| 001-33357 |
| 10.9 |
| January 8, 2007 | |||
10.2 |
|
| 10-K |
| 001-33357 |
| 10.21 |
| March 17, 2008 | |||
10.3†† |
|
| 10-Q |
| 001-33357 |
| 10.1 |
| May 14, 2021 | |||
10.4†† |
|
| 10-Q |
| 001-33357 |
| 10.2 |
| May 14, 2021 | |||
10.5†† | Binding Term Sheet between Protalix Ltd. and Chiesi Farmaceutici S.p.A. | 10-Q |
| 001-33357 | 10.3 |
| May 14, 2021 | |||||
10.6†† |
|
| 10-Q/A |
| 001-33357 |
| 10.1 |
| December 11, 2015 | |||
10.7†† |
|
| 10-K |
| 001-33357 |
| 10.16 |
| March 6, 2018 | |||
79
10.8†† |
|
| 10-Q |
| 001-33357 |
| 10.1 |
| November 7, 2018 | |||
10.9† |
|
| 8-K |
| 001-33357 |
| 10.1 |
| May 21, 2019 | |||
10.10† |
|
| 8-K |
| 001-33357 |
| 10.1 |
| July 29, 2019 | |||
10.11 | 8-K | 001-33357 |
| 10.1 |
| March 12, 2020 | ||||||
10.12† | Amended and Restated Pro BioTherapeutics, Inc. 2006 Stock Incentive Plan, as amended | 10-Q | 001-33357 |
| 10.1 | August 7, 2023 | ||||||
10.13† | Employment Agreement with Yael Hayon, Ph.D. dated June 7, 2020 | 8-K | 001-33357 |
| 10.1 |
| June 8, 2020 | |||||
10.14 | 8-K |
| 001-33357 |
| 10.1 | August 12, 2021 | ||||||
10.15 | 8-K |
| 001-33357 |
| 10.1 | August 26, 2021 | ||||||
10.16 | 8-K |
| 001-33357 |
| 10.2 | August 26, 2021 | ||||||
10.17†† | 10-Q |
| 001-33357 |
| 10.1 | November 14, 2022 | ||||||
10.18†† | Letter Agreement dated August 29, 2022 from Chiesi Farmaceutici S.p.A to Protalix Ltd. | 10-Q |
| 001-33357 | 10.2 | November 14, 2022 | ||||||
21.1 |
|
| 10-K |
| 001-33357 |
| 21.1 |
| February 26, 2010 | |||
80
23.1 |
|
|
|
|
|
| X | |||||
24.1 | Power of Attorney (included on the signature page to this Annual Report on Form 10-K) | X | ||||||||||
31.1 |
|
|
|
|
|
| X | |||||
31.2 |
|
|
|
|
|
| X | |||||
|
|
|
|
|
|
| ||||||
32.1 |
|
|
|
|
|
| X | |||||
|
|
|
|
|
|
| ||||||
32.2 |
|
|
|
|
|
| X | |||||
97.1 | Policy Relating to Recovery of Erroneously Awarded Compensation | X | ||||||||||
101.INS |
| XBRL INSTANCE DOCUMENT - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document |
|
|
|
|
| X | ||||
|
|
|
|
|
|
| ||||||
101.SCH |
| XBRL SHEMA FILE |
|
|
|
|
| X | ||||
|
|
|
|
|
|
| ||||||
101.CAL |
| XBRL CALCULATION FILE |
|
|
|
|
| X | ||||
101.DEF |
| XBRL DEFINITION FILE |
|
|
|
|
| X | ||||
|
|
|
|
|
|
| ||||||
101.LAB |
| XBRL LABEL FILE |
|
|
|
|
| X | ||||
|
|
|
|
|
|
| ||||||
101.PRE |
| XBRL PRESENTATION FILE |
|
|
|
|
| X | ||||
104 | Cover Page Interactive Data File - formatted in Inline XBRL and included as Exhibit 101 | X |
81
† Management contracts or compensation plans or arrangements in which directors or executive officers are eligible to participate.
†† Portions of this exhibit were omitted and have been filed separately with the Secretary of the Securities and Exchange Commission pursuant to the Registrant’s application requesting confidential treatment under Rule 24b-2 of the Exchange Act.
Item 16. Form 10-K Summary
None.
82
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, as of March 14, 2024.
| PROTALIX BIOTHERAPEUTICS, INC. | |
|
| |
| By: | /s/ Dror Bashan |
|
| Dror Bashan |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Dror Bashan and Eyal Rubin, and each of them, as his true and lawful attorneys-in-fact and agents, with full power of substitution and re-substitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming that said attorneys-in-fact and agents, or any of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Signature |
| Title |
| Date |
|
|
|
| |
/s/ Dror Bashan |
| President, Chief Executive Officer |
| March 14, 2024 |
Dror Bashan |
| (Principal Executive Officer) and Director |
|
|
|
|
|
| |
/s/ Eyal Rubin |
| Chief Financial Officer, Treasurer and |
| March 14, 2024 |
Eyal Rubin |
| Secretary (Principal Financial |
|
|
and Accounting Officer) | ||||
|
|
|
| |
/s/ Eliot Richard Forster |
| Chairman of the Board |
| March 14, 2024 |
Eliot Richard Forster, Ph.D. |
|
|
| |
|
|
|
| |
/s/ Amos Bar Shalev |
| Director |
| March 14, 2024 |
Amos Bar Shalev |
|
|
| |
|
|
|
| |
/s/ Shmuel Ben Zvi |
| Director |
| March 14, 2024 |
Shmuel Ben Zvi, Ph.D. |
|
|
| |
|
|
|
| |
/s/ Pol F. Boudes |
| Director |
| March 14, 2024 |
Pol F. Boudes, M.D. |
|
|
| |
|
|
|
| |
/s/ Gwen A. Melincoff |
| Director |
| March 14, 2024 |
Gwen A. Melincoff |
|
|
| |
|
|
|
| |
/s/ Aharon Schwartz |
| Director |
| March 14, 2024 |
Aharon Schwartz, Ph.D. |
|
|
|
83
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED FINANCIAL STATEMENTS
TABLE OF CONTENTS
|
| Page |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM (PCAOB name: | F-2 | |
CONSOLIDATED FINANCIAL STATEMENTS | ||
Consolidated Balance Sheets as of December 31, 2022 and 2023 | F-4 | |
Consolidated Statements of Operations for the years ended December 31, 2021, 2022 and 2023 | F-5 | |
F-6 | ||
Consolidated Statements of Cash Flows for the years ended December 31, 2021, 2022 and 2023 | F-7 | |
F-9 |
F-1

Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Protalix BioTherapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Protalix BioTherapeutics, Inc. and its subsidiaries (the “Company”) as of December 31, 2023 and 2022, and the related consolidated statements of operations, changes in stockholders’ equity (capital deficiency) and cash flows for each of the three years in the period ended December 31, 2023, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023 in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that (i) relates to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
F-2
Recognition and measurements of sales reserve
As discussed in Note 2(k) to the consolidated financial statements revenues from selling goods are recorded in the amount of consideration to which the Company expects to be entitled in exchange for its performance obligation upon transfer of control to the customer. The amount of consideration to which the Company expects to be entitled varies as a result of potential discounts, rebates and allowances. A variable consideration is recorded by the Company concurrently with the satisfaction of its performance obligation to the extent that it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. As of December 31, 2023, the sales reserve for potential discounts, rebates and allowances was $2,861 thousand. The sales reserve is calculated based on expected sales and anticipated net average selling price.
The principal considerations for our determination that performing procedures related to recognition and measurement of the sales reserve is a critical audit matter are the significant judgment by management due to the significant measurement uncertainty involved in developing the sales reserve, as the sales reserve is based on assumptions developed related to expected sales and anticipated net average selling price. This in turn led to significant auditor judgment in performing procedures relating to these assumptions.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included, among others, (i) testing management’s process for determining the estimate; and (ii) testing the completeness, accuracy, and relevance of underlying data used to estimate the reserves.
/s/ Kesselman & Kesselman
Certified Public Accountants (Isr.)
A member of PricewaterhouseCoopers International Limited
March 14, 2024
We have served as the Company’s auditor since 2000.
F-3
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED BALANCE SHEETS
(U.S. dollars in thousands)
December 31, | ||||||
2022 | 2023 | |||||
ASSETS | ||||||
CURRENT ASSETS: | ||||||
Cash and cash equivalents | $ | | $ | | ||
Short-term bank deposits | | | ||||
Accounts receivable – Trade |
| |
| | ||
Other assets |
| |
| | ||
Inventories |
| |
| | ||
Total current assets | $ | | $ | | ||
NON-CURRENT ASSETS: | ||||||
Funds in respect of employee rights upon retirement | $ | | $ | | ||
Property and equipment, net |
| |
| | ||
Deferred income tax asset | | |||||
Operating lease right of use assets |
| |
| | ||
Total assets | $ | | $ | | ||
LIABILITIES AND STOCKHOLDERS' EQUITY (NET OF CAPITAL DEFICIENCY) |
|
|
| |||
CURRENT LIABILITIES: |
|
|
| |||
Accounts payable and accruals: |
|
|
| |||
Trade | $ | | $ | | ||
Other |
| |
| | ||
Operating lease liabilities |
| |
| | ||
Contracts liability |
| |
| |||
Convertible notes | | |||||
Total current liabilities | $ | | $ | | ||
LONG TERM LIABILITIES: |
|
|
| |||
Convertible notes | $ | | ||||
Liability for employee rights upon retirement |
| | $ | | ||
Operating lease liabilities |
| |
| | ||
Total long term liabilities | $ | | $ | | ||
Total liabilities | $ | | $ | | ||
COMMITMENTS | ||||||
STOCKHOLDERS' EQUITY (CAPITAL DEFICIENCY) | ||||||
Common Stock, $ | | | ||||
Additional paid-in capital | | | ||||
Accumulated deficit | ( | ( | ||||
Total stockholders' equity (capital deficiency) | ( | | ||||
Total liabilities and stockholders' equity (net of capital deficiency) | $ | | $ | | ||
The accompanying notes are an integral part of the consolidated financial statements.
F-4
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(U.S. dollars in thousands, except share and per share amounts)
Year Ended December 31, | |||||||||
2021 |
| 2022 |
| 2023 | |||||
REVENUES FROM SELLING GOODS | $ | | $ | | $ | | |||
REVENUES FROM LICENSE AND R&D SERVICES |
| |
| |
| | |||
TOTAL REVENUE | | | | ||||||
COST OF GOODS SOLD |
| ( |
| ( |
| ( | |||
RESEARCH AND DEVELOPMENT EXPENSES |
| ( |
| ( |
| ( | |||
SELLING, GENERAL AND ADMINISTRATIVE EXPENSES |
| ( |
| ( |
| ( | |||
OPERATING INCOME (LOSS) |
| ( |
| ( |
| | |||
FINANCIAL EXPENSES |
| ( |
| ( |
| ( | |||
FINANCIAL INCOME |
| |
| |
| | |||
FINANCIAL EXPENSES, NET |
| ( |
| ( |
| ( | |||
INCOME (LOSS) BEFORE TAXES ON INCOME | ( | ( | | ||||||
TAXES ON INCOME | — | ( | ( | ||||||
NET INCOME (LOSS) FOR THE PERIOD | $ | ( | $ | ( | $ | | |||
EARNINGS (LOSS) PER SHARE OF COMMON STOCK: | |||||||||
BASIC | $ | ( | $ | ( | $ | | |||
DILUTED | $ | ( | $ | ( | $ | | |||
WEIGHTED AVERAGE NUMBER OF SHARES OF COMMON STOCK | |||||||||
USED IN COMPUTING EARNINGS (LOSS) PER SHARE: | |||||||||
BASIC |
| |
| |
| | |||
DILUTED |
| |
| |
| | |||
The accompanying notes are an integral part of the consolidated financial statements.
F-5
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
(CAPITAL DEFICIENCY)
(U.S. dollars in thousands)
|
|
| Additional |
|
|
| ||||||||
Common | Common | Paid–In | Accumulated | |||||||||||
Stock | Stock | Capital | Deficit | Total | ||||||||||
| Number of |
| ||||||||||||
| Shares | Amount | ||||||||||||
Balance at January 1, 2021 |
| | $ | | $ | | $ | ( | $ | ( | ||||
Changes during 2021: |
|
|
|
|
|
|
|
|
| |||||
Issuance of common stock, net of issuance cost | | | | | ||||||||||
Issuance of common stock under the Sales Agreement, net | | | | | ||||||||||
Share-based compensation related to stock options |
| |
|
|
| | ||||||||
Share-based compensation related to restricted stock award |
| |
|
|
| | ||||||||
Exercise of warrants | | * | — | — | ||||||||||
Reacquisition of equity component of convertible notes | ( | ( | ||||||||||||
Equity component of convertible notes, net of transaction costs | | | ||||||||||||
Net loss |
|
|
| ( |
| ( | ||||||||
Balance at December 31, 2021 |
| | $ | | $ | | $ | ( | $ | ( | ||||
Changes during 2022: |
|
|
|
|
|
|
|
|
| |||||
Issuance of common stock under the Sales Agreement, net | | | | | ||||||||||
Share-based compensation related to stock options |
|
|
| |
|
|
| | ||||||
Share-based compensation related to restricted stock award | | | | | ||||||||||
Exercise of warrants | | * | | | ||||||||||
Net loss |
|
|
|
|
| ( |
| ( | ||||||
Balance at December 31, 2022 |
| | $ | | $ | | $ | ( | $ | ( | ||||
Changes during 2023: |
|
|
|
|
|
|
|
|
|
| ||||
Issuance of common stock under the Sales Agreement, net | | | | | ||||||||||
Convertible notes conversions | | | | | ||||||||||
Share-based compensation related to stock options |
|
|
| |
|
|
| | ||||||
Share-based compensation related to restricted stock award | | | | | ||||||||||
Exercise of warrants | | * | | | ||||||||||
Net income |
|
|
|
|
| |
| | ||||||
Balance at December 31, 2023 |
| | $ | | $ | | $ | ( | $ | | ||||
* | Represents an amount less than $1. |
The accompanying notes are an integral part of the consolidated financial statements.
F-6
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(U.S. dollars in thousands)
Year Ended December 31, | |||||||||
2021 |
| 2022 |
| 2023 | |||||
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
|
| ||||
Net income (loss) | $ | ( | $ | ( | $ | | |||
Adjustments required to reconcile net income (loss) to net cash used in operating activities: |
|
|
| ||||||
Share-based compensation |
| |
| |
| | |||
Depreciation |
| |
| |
| | |||
Financial income, net |
| |
| ( |
| ( | |||
Changes in accrued liability for employee rights upon retirement |
| |
| ( |
| | |||
Changes in deferred tax asset | | | ( | ||||||
Loss (gain) on amounts funded in respect of employee rights upon retirement |
| ( |
| |
| ( | |||
Loss (gain) on sale of fixed assets |
| ( |
| | | ||||
Loss on extinguishment of convertible notes | | | | ||||||
Gain on conversions of convertible notes | | | ( | ||||||
Amortization of debt issuance costs and debt discount | | | | ||||||
Changes in operating assets and liabilities: |
|
|
| ||||||
Increase (decrease) in contracts liability |
| |
| ( |
| ( | |||
Increase in accounts receivable-trade and other assets |
| ( |
| ( |
| ( | |||
Changes in operating lease right of use assets, net |
| |
| ( |
| | |||
Decrease (increase) in inventories |
| ( |
| |
| ( | |||
Increase (decrease) in accounts payable and accruals |
| |
| ( |
| | |||
Decrease in other long term liabilities |
| ( |
| |
| | |||
Net cash used in operating activities | $ | ( | $ | ( | $ | ( | |||
CASH FLOWS FROM INVESTING ACTIVITIES: |
|
|
| ||||||
Investment in bank deposits | $ | ( | $ | ( | $ | ( | |||
Proceeds from sale of short-term deposits | | | | ||||||
Purchase of property and equipment | ( | ( | ( | ||||||
Proceeds from sale of property and equipment | | | | ||||||
Decrease in restricted deposit |
| |
| |
| | |||
Amounts paid (funded) in respect of employee rights upon retirement, net |
| ( |
| |
| ( | |||
Net cash provided by (used in) investing activities | $ | | $ | ( | $ | ( | |||
CASH FLOWS FROM FINANCING ACTIVITIES: | |||||||||
Payment for convertible notes redemption and transactions costs | $ | ( | $ | | $ | | |||
Payment for promissory note | ( | | | ||||||
Proceeds from issuance of common stock and warrants, net | | | | ||||||
Proceeds from issuance of common stock under the Sales Agreement, net | | | | ||||||
Exercise of warrants | | | | ||||||
Net cash provided by financing activities | $ | | $ | | $ | | |||
EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS | $ | | $ | ( | $ | ( | |||
NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS |
| |
| ( |
| | |||
BALANCE OF CASH AND CASH EQUIVALENTS AT BEGINNING OF YEAR | |
| |
| | ||||
BALANCE OF CASH AND CASH EQUIVALENTS AT END OF YEAR | $ | | $ | | $ | | |||
The accompanying notes are an integral part of the consolidated financial statements.
F-7
PROTALIX BIOTHERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS (CONTINUED)
(U.S. dollars in thousands)
Year Ended December 31, | |||||||||
2021 | 2022 |
| 2023 | ||||||
SUPPLEMENTARY INFORMATION ON INVESTING AND FINANCING ACTIVITIES NOT INVOLVING CASH FLOWS: | |||||||||
Purchase of property and equipment | $ | | $ | | $ | | |||
Operating lease right of use assets obtained in exchange for new operating lease liabilities | $ | | $ | | $ | | |||
Convertible notes conversions | $ | - | $ | - | $ | | |||
Partial settlement of liability for employee rights upon retirement through transfer of the related funds | $ | - | $ | - | $ | | |||
SUPPLEMENTARY DISCLOSURE ON CASH FLOWS |
|
|
|
|
| ||||
Interest paid | $ | | $ | | $ | | |||
Interest received | $ | | $ | | $ | | |||
As to extinguishment of convertible notes see Note 10.
The accompanying notes are an integral part of the consolidated financial statements.
F-8
NOTE 1 - SIGNIFICANT ACCOUNTING POLICIES
a. General
Protalix BioTherapeutics, Inc. (collectively with its subsidiaries, the “Company”) and its wholly-owned subsidiaries, Protalix Ltd. and Protalix B.V. (collectively, the “Subsidiaries”), are biopharmaceutical companies focused on the development and commercialization of recombinant therapeutic proteins based on the Company’s proprietary ProCellEx® protein expression system (“ProCellEx”). To date, the Company has successfully developed two enzyme replacement therapies (ERTs); Elfabrio® (pegunigalsidase alfa) for the treatment of adult patients with a confirmed diagnosis of Fabry disease and Elelyso® (taliglucerase alfa) for the treatment of adult patients with Gaucher disease. Elfabrio, which the Company referred to as PRX-102 during its development stage, has been approved for marketing in the United States, the European Union, Great Britain and Switzerland. The Company has partnered with Chiesi Farmaceutici S.p.A. (“Chiesi”) for the development and commercialization of Elfabrio.
The Company has licensed the rights to commercialize Elelyso worldwide (other than Brazil) to Pfizer Inc. (“Pfizer”), and in Brazil to Fundação Oswaldo Cruz (“Fiocruz”), an arm of the Brazilian Ministry of Health (the “Brazilian MoH”). Elelyso is marketed as BioManguinhos alfataliglicerase in Brazil.
The Company’s strategy is to develop proprietary recombinant proteins designed to address high, unmet needs in the rare disease space that are therapeutically superior to existing recombinant proteins currently marketed for the same indications. Consistent with this strategy, the Company has a number of product candidates in varying stages of the clinical development process.
On May 5, 2023, the European Commission (“EC”) announced that it had approved the Marketing Authorization Application (“MAA”) for Elfabrio and on May 9, 2023, the U.S. Food and Drug Administration (“FDA”) announced that it had approved the Biologics License Application (“BLA”) for Elfabrio, each for adult patients with a confirmed diagnosis of Fabry disease. Both approvals cover the 1 mg/kg every two weeks dosage. The European Medicines Agency (“EMA”) approval followed the February 2023 adoption of a positive opinion and recommendation of marketing authorization for Elfabrio by the EMA’s Committee for Medicinal Products for Human Use the (“CHMP”). Elfabrio was approved by the FDA with a boxed warning for hypersensitivity reactions/anaphylaxis, consistent with Enzyme Replacement Therapy (ERT) class labeling, and Warnings/Precautions providing guidance on the signs and symptoms of hypersensitivity and infusion-associated reactions seen in the clinical studies as well as treatments to manage such events should they occur. The Warnings/Precautions for membranoproliferative glomerulonephritis (MPGN) alert prescribers to the possibility of MPGN and provide guidance for appropriate patient management. Overall, the FDA review team concluded that in the context of Fabry disease as a rare, serious disease with limited therapeutic options that may not be suitable to all individual patients, the benefit-risk of Elfabrio is favorable for the treatment of adults with confirmed Fabry disease.
The Company has entered into
Elfabrio was the subject of a phase III clinical program studying the drug as a treatment of patients with Fabry disease, a rare, genetic lysosomal disorder. The phase III clinical program included three separate studies, which are referred to as the BALANCE study, the BRIDGE study and the BRIGHT study. The phase III clinical program analyzed two potential dosing regimens: 1 mg/kg every two weeks and 2 mg/kg every four weeks. In addition, the phase III clinical program included two extension studies in which
F-9
subjects that participated in the Company’s phase I/II clinical trials and phase III clinical trials had the opportunity to enroll and continue to be treated with PRX-102. As of March 1, 2023, sponsorship of the two open label extension studies was transferred to Chiesi, which is now administering the extension studies. From time to time, and as Elfabrio is approved for marketing in different jurisdictions, participants withdraw from the open label extension studies. Some of the withdrawals transfer to a commercial setting, others withdraw for other reasons.
The BLA for Elfabrio for the treatment of adult patients with Fabry disease was resubmitted to the FDA on November 9, 2022. An initial BLA for Elfabrio was submitted to the FDA on May 27, 2020 under the FDA’s Accelerated Approval pathway, but resulted in a Complete Response Letter (“CRL”).
The MAA was submitted to the EMA on February 7, 2022, after the October 8, 2021 meeting the Company held, together with Chiesi, with the Rapporteur and Co-Rapporteur of the EMA regarding PRX-102.
The FDA publicly released the internal review documents for Elfabrio (pegunigalsidase alfa-iwxj) injection BLA 761161. These documents provide previously unavailable additional information regarding the basis for the FDA’s May 2023 approval decision. In particular, the FDA determined that substantial evidence of effectiveness for Elfabrio in Fabry patients was established with one adequate and well-controlled study (Study PB-102-F01/02) with confirmatory evidence provided by the BALANCE study (also referred to as Study PB-102-F20). The FDA review team also concluded that the BALANCE study met its primary efficacy endpoint, which assessed the annualized rate of change in eGFR (estimated glomerular filtration rate) over 104 weeks. However, the FDA also determined that the results from the BALANCE study did not support a non-inferiority claim to the comparator product due to the lack of data to support a non-inferiority margin.
The Company continuously evaluates potential strategic marketing partnerships as well as collaboration programs with biotechnology and pharmaceutical companies and academic research institutions. Except with respect to Elfabrio and Elelyso, the Company holds the worldwide commercialization rights to its other proprietary development candidates.
The Company’s product pipeline currently includes, among other candidates:
| (1) | PRX-115, the Company’s plant cell-expressed recombinant PEGylated uricase (urate oxidase) – a chemically modified enzyme to treat severe gout; and |
| (2) | PRX-119, the Company’s plant cell-expressed PEGylated recombinant human DNase I product candidate being designed to elongate half-life in the circulation for NETs-related diseases. |
Obtaining marketing approval with respect to any product candidate in any country is dependent on the Company’s ability to implement the necessary regulatory steps required to obtain such approvals, and demonstrate the safety and efficacy of its product candidates. The Company cannot reasonably predict the outcome of these activities.
On March 21, 2023, the first patient was dosed in the Company’s phase I First-in-Human (“FIH”) clinical trial of PRX-115. As of December 31, 2023,
On July 2, 2021, the Company entered into an At The Market Offering Agreement (the “2021 Sales Agreement”) with H.C. Wainwright & Co., LLC, as the Company’s sales agent (the “Agent”) which was amended on May 2, 2022. Pursuant to the terms of the 2021 Sales Agreement, the Company was able to sell from time to time through the Agent shares of its common stock, par value $
F-10
Agreement it had entered into on October 1, 2020 with BofA Securities, Inc. (“BofA Securities”). During the term of the sales agreement with BofA Securities, the Company sold a total of
During the term of the 2021 Sales Agreement which ended during the quarter ended March 31, 2023, the Company sold a total of
On February 27, 2023, the Company entered into an At The Market Offering Agreement (the “2023 Sales Agreement”) with the Agent. Pursuant to the terms of the 2023 Sales Agreement, the Company may sell, from time to time through the Agent, ATM Shares having an aggregate offering price of up to $
Under each of the Chiesi Ex-US Agreement and the Chiesi US Agreement (collectively, the “Chiesi Agreements”), Chiesi made an upfront payment to Protalix Ltd. of $
On May 13, 2021, the Company signed a binding term sheet with Chiesi pursuant to which the Company and Chiesi amended the Chiesi Agreements in order to provide the Company with near-term capital. Chiesi agreed to make a $
Under the terms of both of the Chiesi Agreements, Protalix Ltd. is required to manufacture all of the Elfabrio drug substance needed under the agreements, subject to certain exceptions, and Chiesi will purchase Elfabrio drug product from Protalix, subject to certain terms and conditions. The consideration for Protalix Ltd. is based on the drug product supplied to Chiesi and the average selling price of the drug product in the relevant territory multiplied by tiered payments as described in the relevant agreement. Under the Chiesi Ex-US Agreement, the price payable to the Company for drug product supplied is based on a range of
On August 29, 2022, the Company entered into a Fill/Finish Agreement (the “F/F Agreement”) and a Letter Agreement (the “Letter Agreement”), in each case with Chiesi. The Company agreed to supply Chiesi with drug substance for pegunigalsidase alfa in connection with the commercial fill/finish services. Under the F/F Agreement, and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide the Company with commercial fill/finish services for pegunigalsidase alfa, including to support the anticipated global launch of pegunigalsidase alfa. The F/F Agreement shall continue in force until December 31, 2025, unless terminated earlier in accordance with
F-11
the terms of the F/F Agreement and the term may be extended by mutual agreement for an additional period of
Since its approval by the FDA, Elelyso has been marketed by Pfizer in accordance with the exclusive license and supply agreement entered into between Protalix Ltd. and Pfizer, which is referred to herein as the Pfizer Agreement. In October 2015, Protalix Ltd. and Pfizer entered into an amended exclusive license and supply agreement, which is referred to herein as the Amended Pfizer Agreement, pursuant to which the Company sold to Pfizer its share in the collaboration created under the Pfizer Agreement for the commercialization of Elelyso. As part of the sale, the Company agreed to transfer its rights to Elelyso in Israel to Pfizer while gaining full rights to it in Brazil. Under the Amended Pfizer Agreement, Pfizer is entitled to all of the revenues, and is responsible for
On June 18, 2013, the Company entered into a Supply and Technology Transfer Agreement (the “Brazil Agreement”) with Fiocruz for BioManguinhos alfataliglicerase. Fiocruz’s purchases of BioManguinhos alfataliglicerase to date have been significantly below certain agreed-upon purchase milestones and, accordingly, the Company has the right to terminate the Brazil Agreement. Notwithstanding the termination right, the Company is, at this time, continuing to supply BioManguinhos alfataliglicerase to Fiocruz and patients continue to be treated with BioManguinhos alfataliglicerase in Brazil.
Because the Company’s operations are conducted in the State of Israel, the business and operations may be directly affected by economic, political, geopolitical and military conditions in Israel. In October 2023, Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military targets. Hamas also launched extensive rocket attacks on Israeli population and industrial centers located along Israel’s border with the Gaza Strip and in other areas within the State of Israel attacking a number of civilian and military targets while simultaneously launching extensive rocket attacks on the Israeli population and industrial centers. At the same time, clashes between Israel and Hezbollah in Lebanon have increased. In response, Israel’s security cabinet declared war against the Hamas and a military campaign against these terrorist organizations commenced in parallel to their continued rocket and terror attacks. Moreover, the attacks by Hamas and Hezbollah, and Israel’s defensive measures, may result in a greater regional conflict. It is currently not possible to predict the duration or severity of the ongoing conflict or its effects on the Company’s business, operations and financial conditions. The ongoing conflict is rapidly evolving and developing, and could disrupt certain of the Company’s business and operations, among others. As of the issuance of these financial statements, the impact of the war has not had a material adverse effect on the Company’s operations.
The Company expects to continue to incur significant expenditures in the near future due to research and developments efforts with respect to its product candidates. See also Note 10 with regards to financial covenants under the terms of the Company’s outstanding
b. Basis of presentation
The Company’s financial statements have been prepared in accordance with generally accepted accounting principles in the United States (“U.S. GAAP”).
F-12
c. Use of estimates in the preparation of financial statements
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results may differ from those estimates. As applicable to these consolidated financial statements, the most significant estimates relate to the assessment of sales reserves and valuation allowances.
d. Functional currency
The dollar is the currency of the primary economic environment in which the operations of the Company and its Subsidiaries are conducted. The Company’s revenues are derived in dollars. Most of the Company’s expenses and capital expenditures are incurred in dollars, and the major source of the Company’s financing has been provided in dollars.
Transactions and balances originally denominated in dollars are presented at their original amounts. Balances in non-dollar currencies are translated into dollars using historical and current exchange rates for non-monetary and monetary balances, respectively. For non-dollar transactions and other items (stated below) reflected in the statements of operations, the following exchange rates are used: (i) for transactions – exchange rates at the transaction dates or average rates; and (ii) for other items (derived from non-monetary balance sheet items such as depreciation and amortization, etc.) – historical exchange rates. Currency translation gains and losses are recorded as financial income or expenses, as appropriate.
e. Cash equivalents
The Company considers all short-term, highly liquid investments, which include short-term bank deposits with original maturities of three months or less from the date of purchase, that are not restricted as to withdrawal or use and are readily convertible to known amounts of cash, to be cash equivalents.
f. Accounts Receivables
Accounts receivable have been reduced by an allowance for credit losses. The Company maintains the allowance for estimated losses resulting from the inability of the Company’s customers to make required payments. The allowance represents the current estimate of lifetime expected credit losses over the remaining duration of existing accounts receivable considering current market conditions and supportable forecasts when appropriate. The estimate is a result of the Company’s ongoing evaluation of collectability, customer creditworthiness, historical levels of credit losses and future expectations. As of December 31, 2023 and 2022, the allowance was negligible.
No write-off activity and recoveries for the periods presented were recognized.
g. Inventories
Inventories are valued at the lower of cost or net realizable value. Cost of raw and packaging materials and purchased products is determined using the “moving average” basis.
Cost of finished products is determined as follows: the value of the raw and packaging materials component is determined primarily using the “moving average” basis; the value of the labor and overhead component is determined on an average basis over the production period.
Inventory is written down for estimated obsolescence based upon management assumptions about future demand and market conditions.
F-13
h. Property and equipment
1. Property and equipment are stated at cost, net of accumulated depreciation and amortization.
2. The Company’s assets are depreciated by the straight-line method on the basis of their estimated useful lives as follows:
| Years | |
Laboratory equipment |
| |
Furniture |
| |
Computer equipment |
|
Leasehold improvements are amortized by the straight-line method over the shorter of (i) the expected lease term and (ii) the estimated useful life of the improvements.
i. Impairment in value of long-lived assets
The Company tests long-lived assets for impairment if an indication of impairment exists. If the sum of expected future cash flows of definite life assets (undiscounted) is less than the carrying amount of such assets, the Company recognizes an impairment loss, and writes down the assets to their estimated fair values.
j. Income taxes
1. Deferred income taxes
Deferred taxes are determined utilizing the assets and liabilities method based on the estimated future tax effects of the differences between the financial accounting and tax bases of assets and liabilities under the applicable tax laws. Deferred tax balances are computed using the tax rates expected to be in effect when those differences reverse. A valuation allowance in respect of deferred tax assets is provided if, based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
2. Uncertainty in income taxes
Tax benefits recognized in the financial statements are those that the Company’s management deems at least more likely than not to be sustained, based on technical merits. The amount of benefits recorded for these tax benefits is measured as the largest benefit the Company’s management deems more likely than not to be sustained. Liabilities relating to uncertain tax positions are classified as current in the consolidated balance sheets to the extent the Company anticipates making payments within one year.
k. Revenue Recognition
Under Accounting Standards Codification (“ASC”), Topic 606, Revenue from Contracts with Customers, a contract with a customer exists only when: the parties to the contract have approved it and are committed to perform their respective obligations, the Company can identify each party’s rights regarding the distinct goods or services to be transferred (“performance obligations”), the Company can determine the transaction price for the goods or services to be transferred, the contract has commercial substance and it is probable that the Company will collect the consideration to which it will be entitled in exchange for the goods or services that will be transferred to the customer.
F-14
Revenues are recorded in the amount of consideration to which the Company expects to be entitled in exchange for performance obligations upon transfer of control to the customer.
1. Revenues from selling products
The Company recognizes revenues from selling goods at a point in time when control over the product is transferred to customers (upon delivery), at the net selling price, which reflects reserves for variable consideration, potential discounts and allowances. The sales reserve is calculated based on expected sales and the anticipated net average selling price.
The transaction price is the consideration to which the Company expects to be entitled from the customer. The consideration promised in a contract with the Company’s customers may include fixed amounts and variable amounts. The Company estimates the variable consideration and includes it in the transaction price using the most likely outcome method, and only to the extent the variable consideration is probable that a significant reversal of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue.
2. Revenues from Chiesi Agreements
The Company has identified
The Company determined that the license together with the research and development services should be combined into a single performance obligation since Chiesi cannot benefit from the license without the research and development services. The research and development services are highly specialized and are dependent on the supply of the drug.
The future manufacturing was contingent on regulatory approvals of the drug and the Company deems these services to be separately identifiable from other performance obligations in the contract. Manufacturing services post-regulatory approval are not interdependent or interrelated with the license and research and development services. Following the regulatory approvals for Elfabrio received in May 2023, the Company started recognizing revenue from manufacturing. See also revenue from selling products above.
The transaction price was comprised of fixed consideration and variable consideration (capped research and development reimbursements). Under ASC 606, the consideration to which the Company would be entitled upon the achievement of contractual milestones, which are contingent upon the occurrence of future events, are a form of variable consideration. The Company estimates variable consideration using the most likely amount method. Amounts included in the transaction price are recognized only when it is probable that a significant reversal of cumulative revenues will not occur. Prior to recognizing revenue from variable consideration, the Company uses significant judgment to determine the probability of significant reversal of such revenue. Following the approval of Elfabrio by the FDA, the Company received a milestone payment equal to $
Since the customer benefits from the research and development services as the entity performs the service, revenue from granting the license and the research and development services is recognized over time using the cost-to-cost method.
Revenue from additional research and development services ordered by Chiesi, is recognized over time using the cost-to-cost method.
F-15
3. Revenue from R&D services
Revenue from the research and development services is recognized over time using the cost-to-cost method since the customer benefits from the research and development services as the entity performs the service.
l. Research and development costs
Research and development costs are expensed as incurred and consist primarily of personnel, subcontractors and consultants (mainly in connection with clinical trials), facilities, equipment and supplies for research and development activities. In connection with purchases of assets, amounts assigned to intangible assets to be used in a particular research and development project that have no alternative future use are charged to research and development costs at the purchase date. Costs incurred for performing research and development services are included in research and development expenses.
m. Concentration of credit risks and trade receivable
Financial instruments that potentially subject the Company to concentration of credit risk consist principally of bank deposits and account receivables - trade. The Company’s deposits are instruments with highly rated financial institutions, mainly in Israeli banks, and, as a matter of policy, limits the amounts of credit exposure to any one financial institution. The Company has not experienced any credit losses in these accounts and does not believe it is exposed to any significant credit risk on these instruments. The Company’s trade receivables represent amounts to be received from its customers. The Company does not require its customers to post collateral with respect to receivables.
As of December 31, 2022, the accounts receivables balance was composed of $
n. Share-based compensation
The Company accounts for share-based payment awards classified as equity awards, including stock-based option awards and restricted stock, using the grant-date fair value method. The fair value of share-based payment transactions is recognized as an expense over the requisite service period.
The Company calculates the fair value of stock-based option awards on the date of grant using the Black-Scholes option pricing model. This option pricing model requires estimates as to the option’s expected term and the price volatility of the underlying stock.
The Company measures compensation expense for restricted stock based on the market value of the underlying stock at the date of grant.
The Company elected to recognize compensation cost for awards to employees, consultants and other service providers with only service conditions that has a graded vesting schedule using the accelerated method.
The Company elects to account for forfeitures as they occur.
o. Net earnings (loss) per share
Basic earnings (loss) per share is calculated by dividing the net income (loss) by the weighted average number of shares of Common Stock outstanding during each period.
F-16
In computing diluted earnings per share, basic earnings per share are adjusted to take into account the potential dilution that could occur upon: (i) the exercise of options granted under employee stock compensation plans using the treasury stock method; (ii) the exercise of warrants using the treasury stock method; and (iii) the conversion of the convertible notes using the “if-converted” method.
p. Convertible notes
The outstanding convertible notes are accounted for using the guidance set forth in the Financial Accounting Standards Board (“FASB”) ASC 815 requiring that the Company determine whether the embedded conversion option must be separated and accounted for separately. ASC 470-20 regarding debt with conversion and other options requires the issuer of a convertible debt instrument that may be settled in cash upon conversion to separately account for the liability (debt) and equity (conversion option) components of the instrument in a manner that reflects the issuer’s nonconvertible debt borrowing rate. The Company’s outstanding
Issuance costs regarding the issuance of the 2021 Notes, as well as the debt discount and debt issuance costs from the issuance of the 2024 Notes, were deferred and amortized over the applicable convertible notes period using the effective interest rate.
As of December 31, 2023, a total of $
q. Leases
Leases are classified as finance or operating, with classification affecting the pattern and classification of expense recognition in the statement of operations. The Company determines if an arrangement is a lease at inception. Lease classification is governed by five criteria in ASC 842-10-25-2. If any of these five criteria is met, the Company classifies the lease as a finance lease. Otherwise, the Company classifies the lease as an operating lease. The Company does not have any finance leases.
Operating leases are included in operating lease right-of-use (“ROU”) assets and operating lease liabilities in the consolidated balance sheets.
ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at the commencement date based on the present value of lease payments over the lease term. The Company uses its incremental borrowing rate based on the information available at the commencement date to determine the present value of the lease payments.
The Company elected the short-term lease recognition exemption for all leases with a term shorter than 12 months. This means, for those leases, the Company does not recognize ROU assets or lease liabilities.
Lease terms include options to extend or terminate the lease when it is reasonably certain that the Company will either exercise or not exercise the option to renew or terminate the lease. The Company recognizes lease expenses over the lease term on a straight-line basis.
The Company applies the portfolio approach to account for the operating lease ROU assets and liabilities for certain car leases and incremental borrowing rates.
F-17
r. New accounting pronouncements
Recently issued accounting pronouncements, not yet adopted
In August 2020, the FASB issued ASU 2020-06 “Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging – Contracts in Entity’s Own Equity (Subtopic 815 – 40).” This guidance simplifies the accounting for certain financial instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. The amendments to this guidance are effective for fiscal years beginning after December 15, 2023, and interim periods within those fiscal years. Early adoption is permitted, but no earlier than fiscal years beginning after December 15, 2020, including interim periods within those fiscal years. The Company is currently evaluating the impact of the adoption of this standard on its consolidated financial statements.
In December 2023, the FASB issued ASU 2023-09 “Income Taxes (Topic 740): Improvements to Income Tax Disclosures.” This guidance is intended to enhance the transparency and decision-usefulness of income tax disclosures. The amendments in ASU 2023-09 address investor requests for enhanced income tax information primarily through changes to disclosure regarding rate reconciliation and income taxes paid both in the U.S. and in foreign jurisdictions. ASU 2023-09 is effective for fiscal years beginning after December 15, 2024 on a prospective basis. Early adoption is permitted, with the option to apply the standard retrospectively. The Company is currently evaluating this guidance to determine the impact it may have on its consolidated financial statements disclosures.
In November 2023, the FASB issued ASU 2023-07 “Segment Reporting: Improvements to Reportable Segment Disclosures.” This guidance expands public entities’ segment disclosures primarily by requiring disclosure of significant segment expenses that are regularly provided to the chief operating decision maker and included within each reported measure of segment profit or loss, an amount and description of its composition for other segment items, and interim disclosures of a reportable segment’s profit or loss and assets. Public entities with a single reportable segment are required to provide the new disclosures and all the disclosures required under ASC 280. The guidance is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024, with early adoption permitted. The amendments are required to be applied retrospectively to all prior periods presented in an entity’s financial statements. The Company is currently evaluating this guidance to determine the impact it may have on its consolidated financial statements and related disclosures.
NOTE 2 - COMMERCIALIZATION AGREEMENTS
| a. | In October 2015, the Company entered into the Amended Pfizer Agreement with Pfizer that amended and restated the Pfizer Agreement of November 30, 2009. Pursuant to the amendment, the Company granted Pfizer an exclusive license in the entire world, including Israel but excluding Brazil. Pfizer acquired all the information, knowledge and permission to manufacture and sell Elelyso. |
Protalix Ltd. also agreed to provide Pfizer with:
Promissory note – as of the date of the amendment, the Company issued to Pfizer a $
F-18
Operation. The promissory note was subsequently amended. On March 29, 2021, the Company paid approximately $
b. In October 2017, Protalix Ltd. entered into the Chiesi Ex-U.S. Agreement with respect to the commercialization of pegunigalsidase alfa (hereafter – the drug) for the treatment of Fabry disease. Under the terms of the Chiesi Agreement, Protalix Ltd. granted to Chiesi exclusive licensing rights for the commercialization of the drug for all markets outside of the United States. At the effective date, Protalix Ltd. had maintained the exclusive commercialization rights to the drug in the United States, which rights were subsequently granted to Chiesi in July 2018.
Protalix Ltd. will be mainly responsible for (i) continuing the development of the drug until a regulatory approval is granted and (ii) manufacture and supply the drug to Chiesi, based on Chiesi’s requests.
The consideration consists of the following:
| 1. | Upfront, non-refundable payment of $ |
| 2. | Additional payments of up to $ |
| 3. | Payments for additional studies, as may be approved from time to time by Chiesi. |
| 4. | Milestone payments of up to $ |
| 5. | Additional payments as consideration for the supply of the drug. The payment will vary from |
| 6. | Protalix Ltd. will be the sole manufacturer of the drug. |
Chiesi does not have sublicensing rights (except for certain territories).
In July 2018, Protalix Ltd. entered into the Chiesi U.S. Agreement with respect to the commercialization of the drug for the treatment of Fabry disease. Under the terms of the Chiesi U.S. Agreement, Protalix Ltd. granted to Chiesi exclusive licensing rights for the commercialization of the drug for all markets in the United States. Protalix Ltd. will be mainly responsible for (i) continuing the development of the drug until a regulatory approval is granted, (ii) continuing certain clinical development efforts in relation to the drug after a regulatory approval is granted and (iii) manufacture and supply the drug to Chiesi, based on Chiesi’s requests.
The consideration consists of the following:
| 1. | Upfront, non-refundable payment of $ |
| 2. | Additional payments of up to $ |
| 3. | Payments for additional studies, as may be approved from time to time by Chiesi. |
| 4. | Milestone payments of up to $ |
| 5. | Additional payments as consideration for the supply of the drug. The payment will vary from |
| 6. | Protalix will be the sole manufacturer of the drug. |
F-19
Chiesi does not have sublicensing rights.
As of December 31, 2023, the Company has received, or is entitled to receive, the following payments from Chiesi:
| 1. | Upfront payments equal to $ |
| 2. | Payments equal to $ |
| 3. | Payments equal to approximately $ |
| 4. | Payments equal to $ |
c. On June 18, 2013, Protalix Ltd. entered into the Brazil Agreement with Fiocruz for BioManguinhos. Fiocruz’s purchases of BioManguinhos alfataliglicerase to date have been significantly below certain agreed upon purchase milestones and, accordingly, the Company has the right to terminate the Brazil Agreement. Notwithstanding, the Company is, at this time, continuing to supply BioManguinhos alfataliglicerase to Fiocruz under the Brazil Agreement, and patients continue to be treated with BioManguinhos alfataliglicerase in Brazil.
NOTE 3 - PROPERTY AND EQUIPMENT
a. Composition of property and equipment grouped by major classifications is as follows:
December 31, | ||||||
(U.S. dollars in thousands) |
| 2022 |
| 2023 | ||
Laboratory equipment | $ | | $ | | ||
Furniture and computer equipment |
| |
| | ||
Leasehold improvements |
| |
| | ||
$ | | $ | | |||
Less – accumulated depreciation and amortization |
| ( |
| ( | ||
$ | | $ | | |||
b. Depreciation in respect of property and equipment totaled approximately $
NOTE 4 - INVENTORIES
| a. | Inventories at December 31, 2022 and 2023 consisted of the following: |
December 31, | ||||||
(U.S. dollars in thousands) | 2022 | 2023 | ||||
Raw materials | $ | | $ | | ||
Work in progress |
| | | |||
Finished goods |
| | | |||
Total inventory | $ | | $ | | ||
b. During the years ended December 31, 2021, 2022 and 2023, the Company recorded approximately $
F-20
NOTE 5 - LIABILITY FOR EMPLOYEE RIGHTS UPON RETIREMENT
The Israeli Subsidiary is required to make a severance payment upon dismissal of an employee or upon termination of employment in certain circumstances. The severance pay liability to the employees (based upon length of service and the latest monthly salary - one month’s salary for each year employed) is recorded on the Company’s balance sheets under “Liability for employee rights upon retirement.” The liability is recorded as if it were payable at each balance sheet date on an undiscounted basis.
The liability is funded in part from the purchase of insurance policies or by the establishment of pension funds with dedicated deposits in the funds. The amounts used to fund these liabilities are included in the Company’s balance sheets under “Funds in respect of employee rights upon retirement.” These policies are the Company’s assets. However, under labor agreements and subject to certain limitations, any policy may be transferred to the ownership of the individual employee for whose benefit the funds were deposited. In the years ended December 31, 2021, 2022 and 2023, the Company deposited approximately $
In accordance with the current employment agreements with certain employees, the Company makes regular deposits with certain insurance companies for accounts controlled by each applicable employee in order to secure the employee’s rights upon retirement. The Company is fully relieved from any severance pay liability with respect to each such employee after it makes the payments on behalf of the employee. The liability accrued in respect of these employees and the amounts funded, as of the respective agreement dates, are not reflected in the Company’s balance sheets, as the amounts funded are not under the control and management of the Company and the pension or severance pay risks have been irrevocably transferred to the applicable insurance companies (the “Contribution Plans”).
The amounts of severance pay expenses were approximately $
The Company expects to contribute approximately $
During the five-year period following December 31, 2023, the Company expects
NOTE 6 - COMMITMENTS
a. Royalty Commitments
The Company is obligated to pay royalties to the National Authority for Technological Innovation (“NATI”) on proceeds from the sale of products developed from research and development activities that were funded, partially, by grants from NATI or its predecessor, the Office of the Israeli Innovation Authority (IIA). At the time the grants were received, successful development of the related projects was not assured.
In the case of failure of a project that was partly financed as described above, the Company is not obligated to pay any such royalties or repay funding received from NATI or the IIA.
F-21
Under the terms of the applicable funding arrangements, royalties of
Royalty expenses to NATI or the IIA are included in the statement of operations as a component of the cost of revenues and were approximately $
At December 31, 2022 and 2023, the maximum total royalty amount payable by the Company under these funding arrangements is approximately $
b. Subcontracting Agreements
The Company has entered into sub-contracting agreements with several clinical providers and consultants in Israel, the United States and certain other countries in connection with its primary product development process. As of December 31, 2023, total commitments under said agreements were approximately $
c. Fill/Finish Agreement
On August 29, 2022, the Company entered into the F/F Agreement and the Letter Agreement. The Company agreed to supply Chiesi with drug substance for pegunigalsidase alfa and, following relevant technology and technical information transfer activities, Chiesi has agreed, among other things, to provide the Company with commercial fill/finish services for pegunigalsidase alfa, including to support the anticipated global launch of pegunigalsidase alfa. The F/F Agreement did not change the terms and conditions of the Chiesi Agreements. The F/F Agreement shall continue in force until December 31, 2025, unless terminated earlier in accordance with the terms of the F/F Agreement and the term may be extended by mutual agreement for an additional period of
NOTE 7 - OPERATING LEASES
The Company is a party to several lease agreements for its facilities, the latest of which has been extended until 2026. The Company has the
The Company entered into several
F-22
The following table sets forth data regarding the Company’s operating leases for the years ended December 31, 2021, 2022 and 2023:
Year ended December 31, | ||||||||||
(U.S. dollars in thousands) | 2021 | 2022 | 2023 | |||||||
Operating lease costs | $ | | $ | | $ | | ||||
Cash paid for amounts included in the measurement of lease liabilities | | | | |||||||
Weighted average remaining lease term (in years) | ||||||||||
Weighted average discount rate | | % | | % | | % | ||||
The following table sets forth a maturity analysis of the Company’s operating lease liabilities as of December 31, 2023:
(U.S. dollars in thousands) |
| December 31, 2023 | |
2024 | $ | | |
2025 | $ | | |
2026 | $ | | |
2027 | $ | | |
2028 and thereafter | $ | | |
Total undiscounted cash flows | $ | | |
Less: imputed interest | $ | | |
Present value of operating lease liabilities | $ | | |
NOTE 8 - REVENUE
The following table summarizes the Company’s disaggregation of revenues:
Year Ended December 31, | |||||||||
(U.S. dollars in thousands) | 2021 | 2022 |
| 2023 | |||||
Pfizer | $ | | $ | | $ | | |||
Brazil | $ | | $ | | $ | | |||
Chiesi | | $ | | $ | | ||||
Total revenues from selling goods | $ | | $ | | $ | | |||
Revenues from license and R&D services | $ | | $ | | $ | | |||
During the year ended December 31, 2021, and following the CRL received from the FDA and other understandings with Chiesi, the Company changed its estimate for total costs expected to be incurred until satisfying the performance obligation under the Chiesi Agreements. This resulted in reduced revenues recognized in respect of this performance obligation in 2021.
NOTE 9 - SHARE CAPITAL
a. Authorized Capital
On June 28, 2023, the Company held its 2023 Annual Meeting of Stockholders, which was adjourned and reconvened on July 13, 2023 (the “Annual Meeting”). At the Annual Meeting, the Company’s stockholders, among other matters, approved an amendment to the Company’s Certificate of Incorporation, as amended, to increase the number of shares of Common Stock authorized for issuance from
F-23
to
b.Rights of the Company’s Common Stock
The Company’s Common Stock is listed on the NYSE American. The Company voluntarily delisted its Common Stock from the Tel Aviv Stock Exchange effective March 22, 2023. Each share of Common Stock is entitled to
c. Stock based compensation
On December 14, 2006, the Board of Directors adopted the Protalix BioTherapeutics, Inc. 2006 Stock Incentive Plan, as amended (the “Plan”). The Plan has since been amended to, among other things, increase the number of shares of Common Stock available under the Plan to
As of December 31, 2023,
The vesting period of the outstanding options and restricted shares is generally or
For purposes of determining the fair value of the options and restricted stock granted to employees and non-employees, the Company’s management uses the fair value of the Common Stock. The fair value of options granted for both employees and directors is estimated at the date of grant using the Black-Scholes option-pricing model.
1. Options and restricted stock granted to employees and directors:
A summary of share option plans, and related information, under all of the Company’s equity incentive plans for the year ended December 31, 2023, and the effect of share-based compensation on the statement of operations for the year ended December 31, 2023, is as follows:
a) | Options granted to employees and directors: |
Year ended December 31, 2023 | |||||
|
| Weighted | |||
| Number | average | |||
| of | exercise | |||
| options | price | |||
Outstanding at beginning of year |
| | $ | | |
Changes during the year: |
|
|
|
| |
Granted |
| |
| | |
F-24
Forfeited and expired |
| |
| | |
Outstanding at end of year |
| | $ | | |
Exercisable at end of year |
| | $ | |
b) | Restricted stock granted to employees: |
Year Ended December 31, 2023 | |||
Number of Restricted Stock | |||
Outstanding at beginning of year | |||
Changes during the year: | |||
Granted | |||
Vested | |||
Non vested at end of year |
c) | The following table summarizes information concerning outstanding and exercisable options as of December 31, 2023: |
December 31, 2023 | ||||||||
Options outstanding | Options exercisable | |||||||
| Number of |
| Weighted |
|
| Weighted | ||
options | average | average | ||||||
outstanding | remaining | Number of | remaining | |||||
Exercise | at end of | contractual | options | contractual | ||||
prices | year | life | exercisable | life | ||||
$ | | | ||||||
$ | | | ||||||
$ |
| |
|
| |
| ||
$ |
| |
|
| |
| ||
| |
| | |||||
As of December 31, 2023, the aggregate intrinsic value of all the outstanding options and exercisable options was approximately $
d) | The fair value of each option granted during 2021, 2022 and 2023 for both employees and directors is estimated at the date of grant using the Black-Scholes option-pricing model. The following weighted average assumptions were applied in determining the options’ fair value on their grant date: |
Year Ended December 31, | |||||||
2021 | 2022 | 2023 | |||||
Weighted average grants date fair value (USD) | | | | ||||
Exercise price (USD) | | |
| | |||
Risk free rate | | % | | % | | % | |
Volatility | | % | | % | | % | |
Dividend yield | % | % | % | ||||
Expected life (Years) |
| ||||||
e) | The total unrecognized compensation cost of employee stock options at December 31, 2023 is approximately $ |
F-25
During the three years ended December 31, 2023, there were
The total vesting-date value of equity classified restricted stock vested during the year ended December 31, 2023 was $
f) | The following table illustrates the effect of share-based compensation on the statement of operations: |
Year ended December 31, | |||||||||
(U.S. dollars in thousands) |
| 2021 |
| 2022 |
| 2023 | |||
Cost of goods sold | $ | | $ | | $ | | |||
Research and development expenses | | | | ||||||
Selling, general and administrative expenses |
| |
| |
| | |||
$ | | $ | | $ | | ||||
d. Private and 144A Offerings
On June 7, 2021, the Company issued
During the year ended December 31, 2022, the Company issued
During the year ended December 31, 2023, the Company issued (i)
During the year ended December 31, 2023, the Company issued, in the aggregate,
e. At-the-Market (ATM) Offering
On July 2, 2021, the Company entered into the 2021 Sales Agreement pursuant to which the Company was able to sell, from time to time through the Agent, shares of Common Stock having an aggregate offering price of up to $
The Company has no obligation to sell any shares of Common Stock under the 2023 Sales Agreement, and may at any time suspend sales under the 2023 Sales Agreement or terminate the 2023 Sales Agreement in accordance with its terms. The Agent is entitled to a commission of up to
F-26
During the year ended December 31, 2021, but prior to the termination of the BofA Agreement, the Company sold
During the year ended December 31, 2022, the Company sold, in the aggregate,
During the year ended December 31, 2023, the Company sold, in the aggregate,
f. Public Offering
On February 17, 2021, the Company issued and sold
NOTE 10 - CONVERTIBLE NOTES
a.
In December 2016, the Company issued to certain existing note holders and new purchasers $
The following table sets forth total interest expense recognized related to the 2021 Notes:
Year Ended December 31, | ||||
(U.S. Dollars in thousands) |
| 2021 |
| |
Contractual interest expense | $ | | ||
Debt discount amortization |
| | ||
Loss from extinguishment |
| | ||
Total | $ | | ||
b.
On August 25, 2021, the Company completed exchanges (the “Exchanges”) of a substantial majority of the Company’s outstanding 2021 Notes with certain institutional note holders. The Exchanges involved the exchange of an aggregate of $
F-27
Common Stock), subject to adjustment in certain circumstances, which is based on a
The 2024 Notes were issued pursuant to an indenture (the “2024 Indenture”) entered into between the Company, the guarantors party thereto, The Bank of New York Mellon Trust Company, N.A., as trustee and Wilmington Savings Fund Society, FSB, as collateral agent (the “Collateral Agent”). Interest on the 2024 Notes is payable semi-annually at a rate of
For accounting purposes, as the terms of the 2021 Notes and the 2024 Notes are substantially different, the Exchanges were considered an extinguishment of debt. The Company allocated the fair value of the consideration transferred to the participating note holders between the 2021 Notes and their equity component based on the fair value of the liability component before the extinguishment, and the remainder was allocated to the equity component. As a result, the Company recognized a loss from extinguishment in the statement of operations equal to $
The Company accounted for the 2024 Notes as a liability (debt) and equity component (conversion option) as the convertible notes may be settled wholly or partly in cash, at the option of the Company, when converted. The equity component with respect to the cash conversion feature net of transaction costs of approximately $
Transaction costs in the amount of approximately $
Holders may convert their 2024 Notes at any time. The initial conversion rate for the 2024 Notes is
As of December 31, 2023, a total of $
Prior to the maturity date, the Company may redeem in cash:
a)any or all of the outstanding 2024 Notes if, on or after March 31, 2023, the last reported sale price of the Common Stock for at least
b)all of the 2024 Notes then outstanding if the aggregate principal amount of the 2024 Notes then outstanding is less than
To date, there has been
The 2024 Notes are guaranteed by the Restricted Subsidiaries (as defined in the 2024 Indenture) and are secured by a first-priority security interest in all of the present and after-acquired assets of the Company and each of the Restricted Subsidiaries (the “Collateral”), including, but not limited to, (i)
F-28
Company that is held by the Company or any Restricted Subsidiary, (ii) intellectual property, including all copyrights, copyright licenses, patents, patent licenses, software, trademarks, trademark licenses and trade secrets and other proprietary information, including, but not limited to, domain names, (iii) all cash, deposit accounts, securities accounts, commodities accounts and contract rights, (iv) all real property and leased property, subject to applicable minimum thresholds, as set forth in the 2024 Indenture, and (v) all other tangible and intangibles of the Company and the Guarantors. In connection with the grant of such liens, the Company entered into certain agreements with both the Collateral Agent and Altshuler Shaham Trusts Ltd., as security trustee in Israel. The 2024 Indenture restricts the ability of the Company, the Subsidiaries and any future subsidiaries to make certain investments, including transfers of the Company’s assets that constitute collateral securing the 2024 Notes, in its existing and future foreign subsidiaries, subject to certain exceptions.
Upon (i) the occurrence of a fundamental change (as defined in the 2024 Indenture) or (ii) if the Company calls the 2024 Notes for redemption as described below (either event, a “make-whole fundamental change”) and a holder elects to convert its 2024 Notes in connection with such make-whole fundamental change, the Company will, in certain circumstances, increase the conversion rate by a number of additional shares (the “Additional Shares”). In no event will the conversion rate exceed the maximum conversion rate, which is
If a fundamental change occurs at any time, holders will have the right, at their option, to require the Company to purchase for cash any or all of the 2024 Notes, or any portion of the principal amount thereof, that is equal to $
The Company prepared a valuation of the fair value of the 2024 Notes and 2021 Notes (a Level 3 valuation) as of August 25, 2021. The value was estimated by implementing the binomial model. The liability component was valued based on the Income Approach.
| 2021 Notes | 2024 Notes | ||||
Stock price (USD) |
| | | |||
Expected term |
| | | |||
Risk free rate |
| | % | | % | |
Volatility |
| | % | | % | |
Yield |
| | % | | % |
The following table sets forth total interest expense recognized related to the 2024 Notes:
Year Ended December 31, | ||||||||||
(U.S. dollars in thousands) | 2021 | 2022 |
| 2023 | ||||||
Contractual interest expense | $ | | $ | | $ | | ||||
Amortization of debt issuance costs and debt discount |
| |
| |
| | ||||
Total | $ | | $ | | $ | | ||||
F-29
NOTE 11 - FAIR VALUE MEASUREMENT
The Company discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received from the sale of an asset, or paid to transfer a liability, in an orderly transaction between market participants at the measurement date.
The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
Level 2: Observable prices that are based on inputs not quoted on active markets, but corroborated by market data.
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value.
The fair value of the financial instruments included in the working capital of the Company is usually identical or close to their carrying value.
The fair value of the outstanding $
The Company prepared the valuation of the fair value of the 2024 Notes (a Level 3 valuation) as of December 31, 2023. The value of these notes was estimated by implementing the binomial model. The liability component was valued based on the Income Approach. The following parameters were used:
| 2024 Notes | |||||
Stock price (USD) |
| | ||||
Expected term |
| | ||||
Risk free rate |
| | % | |||
Volatility |
| | % | |||
Yield |
| | % |
NOTE 12 – NET EARNINGS (LOSS) PER SHARE
Basic and diluted net earnings (loss) per share attributable to common stockholders were calculated as follows:
Year Ended December 31, | |||||||||
(In thousands, except share data) | 2021 | 2022 | 2023 | ||||||
Numerator: | |||||||||
Net income (loss) | $ | ( | $ | ( | $ | | |||
Add: | |||||||||
Financial expenses of 2024 Notes* | ( | ||||||||
Net income (loss) for diluted calculation | $ | ( | $ | ( | $ | | |||
Denominator: | |||||||||
F-30
Weighted average shares of Common Stock outstanding for basic calculation | | | | ||||||
Weighted average dilutive effect of 2024 Notes | | ||||||||
Weighted average dilutive effect of stock options | | ||||||||
Weighted average shares of Common Stock outstanding for diluted calculation | | | |
* Financial expenses on 2024 Notes consists of add back of financial expense incurred during the year and inclusion of make-whole interest payments that will be incurred upon conversion.
Diluted loss per share do not include
NOTE 13 - TAXES ON INCOME
| a. | The Company |
Protalix BioTherapeutics, Inc. is taxed according to U.S. tax laws. The Company’s income is taxed in the United States at a rate of up to
The following table summarizes the Company’s taxes on income:
| Year Ended | |||||
(U.S. dollars in thousands) | December 31, 2022 | December 31, 2023 | ||||
Current taxes on income | $ | | $ | | ||
Deferred taxes on income |
| ( | ||||
Total taxes on income | $ | | $ | | ||
The Company had an effective tax rate of
Following the regulatory approvals for Elfabrio in May 2023, the receipt of the $
The TCJA represents fundamental and dramatic modifications to the U.S. tax system. It contains several key tax provisions that impacted the Company including the reduction of the maximum U.S. federal
F-31
corporate income tax rate from 35% to 21%, effective January 1, 2018. Other significant changes under the TCJA includes, among others, a one-time repatriation tax on accumulated foreign earnings, a limitation of NOL deduction to 80% of taxable income, and indefinite carryover of post-2017 NOLs. The TCJA also repealed the corporate alternative minimum tax for tax years beginning after December 31, 2017. Losses generated prior to January 1, 2018 will still be subject to the 20-year carryforward limitation and the alternative minimum tax. Other impacts due to the TCJA included the repeal of the domestic manufacturing deduction, modification of taxation of controlled foreign corporations, a base erosion anti-abuse tax, modification of interest expense limitation rules, modification of limitation on deductibility of excessive executive compensation, and taxation of global intangible low-taxed income.
Modification of interest expense limitation rules under the TCJA provides generally that for taxable years 2019-2022 interest expense deduction shall be limited to 30% of the EBITDA and for taxable years 2022 onwards to 30% of EBIT. Disallowed interest deduction may be carried forward indefinitely. The Company believes that any potential impact (if applicable) of this limitation will be offset by utilization of available NOLs.
Section 174 of the TCJA requires taxpayers to capitalize and amortize research and development expenses for tax years beginning after December 31, 2021. This rule became effective for the Company during the year ended December 31, 2022, and resulted in the capitalization of research and development costs of approximately $
The Company believes that all future profits of its subsidiaries will be indefinitely reinvested or that there is no expectation to distribute any taxable dividends from these subsidiaries. The determination of the amount of the unrecognized deferred tax liability related to the undistributed earnings is estimated as an immaterial amount.
b. Protalix Ltd.
The Company as a “foreign-investment company” measures its results for tax purposes in dollar based on Income Tax Regulations (Bookkeeping Principles of Foreign Invested Companies and of Certain Partnerships and the Determination of Their Taxable Income), 1986. The Israeli Subsidiary is taxed according to Israeli tax laws:
| 1. | Tax rates |
The income of the Israeli Subsidiary, other than income from “Approved Enterprises,” is taxed in Israel at the regular corporate tax rates.
The corporate tax rate was
Capital gain on a sale of assets is subject to capital gain tax according to the corporate tax rate in effect in the year during which the assets are sold.
| 2. | The Law for the Encouragement of Capital Investments, 1959 (the “Encouragement of Capital Investments Law”) |
Under the Encouragement of Capital Investments Law, including Amendment No. 60 to the Encouragement of Capital Investments Law as published in April 2005, by virtue of the “Approved Enterprise” or “Benefited Enterprise” status the Israeli Subsidiary is entitled to various tax benefits as follows:
F-32
a. Reduced tax rates
Income derived from the Approved Enterprise during a 10-year period commencing upon the year in which the enterprise first realizes taxable income is tax exempt, provided that the maximum period to which it is restricted by the Encouragement of Capital Investments Law has not elapsed.
The Israeli Subsidiary has an “Approved Enterprise” plan since 2004 and “Benefited Enterprise” plan since 2009. The period of benefits in respect of the main enterprise of the Company has not yet commenced. The period during which the Company is entitled to benefits in connection with the Benefited Enterprise expired in 2022.
If the Israeli Subsidiary subsequently pays a dividend out of income derived from the “Approved Enterprise” or “Benefited Enterprise” during the tax exemption period, it will be subject to tax on the gross amount distributed (including the company tax on these amounts), at the rate which would have been applicable if such income not been exempted.
b. Accelerated depreciation
The Israeli Subsidiary is entitled to claim accelerated depreciation, as provided by Israeli law, in the first five years of operation of each asset, in respect of buildings, machinery and equipment used by the Approved Enterprise and the Benefited Enterprise.
c. Conditions for entitlement to the benefits
The Israeli Subsidiary’s entitlement to the benefits described above is subject to its fulfillment of conditions stipulated by the law, rules and regulations published thereunder, and the instruments of approval for the specific investment in an approved enterprise. Failure by the Israeli Subsidiary to comply with these conditions may result in the cancellation of the benefits, in whole or in part, and the Subsidiary may be required to refund the amount of the benefits with interest. The Israeli Subsidiary received a final implementation approval with respect to its “Approved Enterprise” from the Investment Center.
d. Amendment of the Law for the Encouragement of Capital Investments, 1959
In recent years, several amendments have been made to the Encouragement of Capital Investments Law which have enabled new alternative benefit tracks, subject to certain conditions.
The Encouragement of Capital Investments Law was amended as part of the Economic Policy Law for the years 2011-2012 (amendment 68 to the Encouragement of Capital Investments Law), which was passed by the Israeli Knesset on December 29, 2010. The amendment sets alternative benefit tracks to those currently in effect under the provisions of the Encouragement of Capital Investments Law. On December 29, 2016, Amendment 73 to the Encouragement of Capital Investments Law was published. This amendment sets new benefit tracks, inter alia, “Preferred Technological Enterprise” and “Special Preferred Technological Enterprise” (the “Capital Investments Law Amendment”).
To date, the Company has elected not to have the Capital Investments Law Amendment apply to the Company.
F-33
c. Tax losses carried forward to future years
As of December 31, 2023 and 2022, the Company had aggregate NOL carry-forwards equal to approximately $
| 1. | The Company |
The Company’s carry-forward NOLs, equal to approximately $
Significant judgment is required in determining any valuation allowance recorded against deferred tax assets. In assessing the need for a valuation allowance, the Company considered all available evidence, including past operating results, the most recent projections for taxable income, and prudent and feasible tax planning strategies. The Company reassesses its valuation allowance periodically and if future evidence allows for a partial or full release of the valuation allowance, a tax benefit will be recorded accordingly.
| 2. | Protalix Ltd. |
At December 31, 2023 and 2022, the Israeli Subsidiary had approximately $
d. Deferred income taxes:
The components of the Company’s net deferred tax assets at December 31, 2022 and 2023 were as follows:
December 31, | ||||||
(U.S. dollars in thousands) |
| 2022 |
| 2023 | ||
In respect of: |
|
|
|
| ||
Research and development expenses | $ | | $ | | ||
Other timing differences |
| ( |
| ( | ||
Net operating loss carry forwards |
| |
| | ||
Valuation allowance |
| ( |
| ( | ||
| |
| | |||
Deferred taxes are computed using the tax rates expected to be in effect when those differences reverse.
e. Reconciliation of the theoretical tax expense to actual tax expense
A reconciliation of the statutory U.S. federal income tax rate of
Year Ended December 31, | |||||||||
(U.S. dollars in thousands) |
| 2021 |
| 2022 |
| 2023 | |||
Tax computed at the statutory U.S. income tax rate |
| $ | ( |
| $ | ( |
| $ | |
Differences in tax rate | - | | ( | ||||||
Statutorily non-deductible expenses | - | | | ||||||
F-34
Change in Valuation allowances | | ( | ( | ||||||
Utilization of carry-forward losses and other temporary items without deferred taxes recognition | - | | | ||||||
Withholding tax | - | - | | ||||||
$ | - | $ | | $ | |
f. Valuation allowance roll-forward
The following table presents the change in the Company’s valuation allowance during the periods presented:
(U.S. dollars in thousands) | |||
Balance at December 31, 2021 | $ | ( | |
Additions to valuation allowance | — | ||
Reductions to valuation allowance | | ||
Balance at December 31, 2022 | $ | ( | |
Balance at December 31, 2022 | $ | ( | |
Additions to valuation allowance | ( | ||
Reductions to valuation allowance | | ||
Balance at December 31, 2023 | $ | ( |
g. Uncertain tax position
Provisions of ASC 740-10, Income Taxes, clarify whether to recognize assets or liabilities for tax positions taken that may be challenged by a tax authority. A reconciliation of the beginning and ending amount of unrecognized tax benefits, is as follows:
(U.S. dollars in thousands) | |||
Balance at December 31, 2021 | $ | - | |
Increase for tax position related to current year | | ||
Balance at December 31, 2022 | $ | | |
Balance at December 31, 2022 | $ | | |
Increase for tax position related to current year | | ||
Balance at December 31, 2023 | $ | |
h. Tax assessments
In accordance with the Income Tax Ordinance, as of December 31, 2023, all of Protalix Ltd.’s tax assessments through tax year 2018 are considered final.
A summary of open tax years by major jurisdiction is presented below:
Jurisdiction: |
| Years: |
Israel |
| |
United States (*) |
|
F-35
(*) | Includes federal, state and local (or similar provincial jurisdictions) tax positions. |
NOTE 14 - SUPPLEMENTARY FINANCIAL STATEMENT INFORMATION
Balance sheets:
December 31, | ||||||
(U.S. dollars in thousands) |
| 2022 |
| 2023 | ||
a. Other assets: |
|
|
|
| ||
Institutions | $ | | $ | | ||
Prepaid expenses |
| |
| | ||
Sundry |
| |
| | ||
$ | | $ | | |||
December 31, | ||||||
(U.S. dollars in thousands) |
| 2022 |
| 2023 | ||
b. Accounts payable and accruals – other: |
|
|
|
| ||
Payroll and related expenses | $ | | $ | | ||
Interest payable |
| |
| | ||
Provision for vacation |
| |
| | ||
Accrued expenses |
| |
| | ||
Royalties payable |
| |
| | ||
Income tax payable | | | ||||
Sales reserves | — | | ||||
Property and equipment suppliers |
| |
| | ||
$ | | $ | | |||
NOTE 15 - RELATED PARTY TRANSACTIONS
| a. | Non-Executive Director Compensation |
Year Ended December 31, | |||||||||
(U.S. dollars in thousands) |
| 2021 |
| 2022 |
| 2023 | |||
Compensation (including share-based compensation) to the non-executive directors | $ | | $ | | $ | | |||
| b. | In September 2023, with the consent of the Audit Committee of the Board of Directors, the Company engaged one of its non-executive directors to advise the Company regarding business development and licensing efforts on a consultancy basis. For the year ended December 31, 2023, the director earned a total of $ |
NOTE 16 - SUBSEQUENT EVENTS
| a. | Since December 31, 2023, the Company has collected approximately $ |
| b. | In January 2024, the Company granted, with the approval of the Company’s compensation committee, |
F-36