UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report: Not applicable
For the transition period from to
Commission file number
(Exact name of Registrant as specified in its charter)
The
(Jurisdiction of incorporation or organization)
The
(Address of principal executive offices)
Tel:
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol(s) |
| Name of each exchange on which registered | |
Securities registered or to be registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act:
None
(Title of Class)
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
Ordinary shares, nominal value € 0.04 per share:
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
◻ Yes ⌧
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
◻ Yes ⌧
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
⌧
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
⌧
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or an emerging growth company. See definition of “large accelerated filer”, “accelerated filer”, and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
◻ Large accelerated filer | ⌧ | ◻ Non-accelerated filer | |||||||
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to section 13(a) of the Exchange Act. ◻
†The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b).
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
◻ U.S. GAAP | ⌧ | ◻ Other |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
◻ Item 17 ◻ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Summary of the Material and Other Risks Associated with Our Business
Our business is subject to numerous material risks and uncertainties that you should be aware of in evaluating our business, including those described in Part I, Item 3.D: “Risk Factors” in this Annual Report on Form 20-F, or this Annual Report. These risks include, but are not limited to, the following:
| ● | We are a biopharmaceutical company with a history of losses. We expect to continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability, which could result in a decline in the market value of our ordinary shares. |
| ● | We will require additional capital to fund our operations and if we fail to obtain the necessary financing, we will not be able to complete the development and commercialization of our product candidates. |
| ● | While we were founded in 2012, we announced our plans to refocus our business on our RNA editing platforms in 2022, and it may make it difficult to assess the future viability of our business and our strategy. |
| ● | Our business depends in part on the success of our product candidates, which are currently in early phases of preclinical development. We cannot be certain that we will be able to successfully complete the clinical development of, obtain regulatory approval for, or successfully commercialize our product candidates. |
| ● | We may not be able to file investigational new drug applications (“INDs”) or IND amendments or similar applications to commence clinical trials of our product candidates on the timelines we expect, and even if we are able to, the U.S. Food and Drug Administration (the “FDA”) or similar foreign regulatory authority may not permit us to proceed. |
| ● | The regulatory approval processes of the FDA, the European Medicines Agency (the “EMA”) and comparable foreign regulatory authorities are lengthy, time-consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our product candidates, our business will be substantially harmed. |
| ● | Failures or delays in the commencement or completion of our preclinical studies or planned clinical trials of our product candidates could result in increased costs to us and could delay, prevent or limit our ability to generate revenue and continue our business. |
| ● | Our RNA technologies are unproven in the disease indications for which they are being developed and tested and may not lead to the development of, or result in, marketable products. |
| ● | Failure to obtain regulatory approval in jurisdictions outside the United States and the European Union would prevent our product candidates from being marketed in those jurisdictions. |
| ● | If a collaborative partner terminates or fails to perform its obligations under an agreement with us, the commercialization of our product candidates, if approved, could be delayed or terminated. |
| ● | Our development and commercialization strategy for our product candidates relies in part upon certain patent rights that we license from third parties, and termination of such license could have a materially adverse effect on our business, financial condition, results of operations and prospects. |
| ● | We rely on third-party manufacturers and suppliers and we intend to rely on third parties to produce preclinical, clinical and commercial supplies of our product candidates. |
| ● | If third parties on which we depend to conduct our preclinical- and clinical studies do not perform as contractually required, fail to satisfy regulatory or legal requirements or miss expected deadlines, our development programs could be delayed with materially adverse effects on our business, financial condition, results of operations and prospects. |
| ● | Our collaboration partnerships with pharmaceutical companies are important to our business. If these companies do not successfully develop drugs pursuant to these agreements, our business could be adversely affected. |
| ● | We license patent rights from third-party owners or licensees and if we fail to comply with our obligations in our intellectual property licenses, we could lose rights that are fundamental to our business. |
| ● | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. |
| ● | We or our licensors or any current or future collaborators or strategic partners may become subject to third party claims or litigation alleging infringement of patents or other proprietary rights or seeking to invalidate patents or other proprietary rights, and we may need to resort to litigation to protect or enforce our patents or other proprietary rights, all of which could be costly, time consuming, delay or prevent the development and commercialization of our product candidates, or put our patents and other proprietary rights at risk. |
| ● | Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business. |
| ● | We face competition from entities that have developed or may develop product candidates for our target indications. If these companies develop technologies or product candidates more rapidly than we do or their technologies, including delivery technologies, are more effective, our ability to develop and successfully commercialize our product candidates may be adversely affected. |
| ● | Even if we are able to commercialize any of our product candidates, the products may not receive coverage and adequate reimbursement from third-party payors, which could harm our business. |
| ● | Any inability to attract and retain qualified key management and technical personnel would impair our ability to implement our business plan. |
| ● | Members of our management board and supervisory board and our principal shareholders and their affiliates have significant control over our company, which will limit other stakeholders’ ability to influence corporate matters and could delay or prevent a change in corporate control. |
| ● | We are a foreign private issuer and, as a result, we are not subject to U.S. proxy rules and are subject to the Securities and Exchange Act of 1934, as amended (the “Exchange Act”) reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company. |
| ● | Certain U.S. holders of our ordinary shares may suffer adverse tax consequences if we are characterized as a passive foreign investment company for U.S. federal income tax purposes. |
| ● | The rights and responsibilities of our shareholders are governed by Dutch law and differ in some important respects from the rights and responsibilities of shareholders under U.S. law. |
The material and other risks summarized above should be read together with the text of the full risk factors discussed in the section entitled “Risk Factors” and the other information set forth in this Annual Report, including our consolidated financial statements and the related notes, as well as in other documents that we file with the Securities and Exchange Commission (the “SEC”). If any such material and other risks and uncertainties occur, our business, prospects, financial condition and results of operations could be materially and adversely affected. The risks summarized above or described in full elsewhere in this Annual Report are not the only risks that we face. Additional risks and uncertainties not currently known to us, or that we currently deem to be immaterial may also materially adversely affect our business, prospects, financial condition and results of operations.
TABLE OF CONTENTS
Page | ||
Summary of the Material and Other Risks Associated with Our Business | ||
4 | ||
5 | ||
7 | ||
Item 1: Identity of Directors, Senior Management and Advisers | 7 | |
7 | ||
7 | ||
7 | ||
7 | ||
7 | ||
7 | ||
57 | ||
7 | ||
57 | ||
58 | ||
89 | ||
89 | ||
89 | ||
89 | ||
90 | ||
96 | ||
3 | ||
101 | ||
101 | ||
E. Critical accounting policies and Significant Judgments and Estimates | 101 | |
101 | ||
101 | ||
104 | ||
106 | ||
111 | ||
111 | ||
111 | ||
111 | ||
113 | ||
113 | ||
113 | ||
113 | ||
114 | ||
1
114 | ||
114 | ||
114 | ||
114 | ||
114 | ||
115 | ||
115 | ||
115 | ||
115 | ||
115 | ||
125 | ||
125 | ||
126 | ||
135 | ||
135 | ||
135 | ||
136 | ||
136 | ||
Item 11: Quantitative and Qualitative Disclosures about Market Risk | 136 | |
Item 12: Description of Securities other that Equity Securities | 137 | |
137 | ||
137 | ||
137 | ||
137 | ||
138 | ||
138 | ||
Item 14: Material Modifications to the Rights of Security Holders and Use of Proceeds | 138 | |
138 | ||
138 | ||
B. Management’s Annual Report on Internal Control over Financial Reporting | 138 | |
C. Attestation Report of the Registered Public Accounting Firm | 139 | |
140 | ||
140 | ||
140 | ||
2
From time to time, we may use our website or our LinkedIn profile at www.linkedin.com/company/proqr-therapeutics/ to distribute material information. Our financial and other material information is routinely posted to and accessible on the Investors & Media section of our website, available at www.ProQR.com. Investors are encouraged to review the Investors & Media section of our website because we may post material information on that site that is not otherwise disseminated by us. Information that is contained in and can be accessed through our website or our LinkedIn page is not incorporated into, and does not form a part of, this Annual Report on Form 20-F.
3
Introduction
This document contains information required for the Annual Report on Form 20-F for the year ended December 31, 2023, of ProQR Therapeutics N.V. (the “Annual Report”). Unless the context specifically indicates otherwise, references in this Annual Report to “ProQR Therapeutics N.V.”, “ProQR Therapeutics”, “ProQR”, “we”, “our”, “ours”, “us”, the “Company” and similar designations refer to ProQR Therapeutics N.V., a company organized under the laws of the Netherlands, and where appropriate, its consolidated subsidiaries.
IFRS Based Information
The audited financial statements as at December 31, 2023 and 2022, and for the years ended December 31, 2023, December 31, 2022 and December 31, 2021, included in the Annual Report have been prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board (“IASB”).
Non-GAAP Information
In presenting and discussing our financial position, operating results and cash flows, management uses certain non-GAAP financial measures. These non-GAAP financial measures should not be viewed in isolation as alternatives to the equivalent IFRS measure and should be used in conjunction with the most directly comparable IFRS measure(s).
Exchange Rates
All references in this Annual Report to “U.S. dollars” or “$” are to the legal currency of the United States, and all references to “€” or “euro” are to the currency of the European Economic and Monetary Union. Our business to date has been conducted primarily in the European Union, and we maintain our books and records in euro. We present our financial statements in euro, which is the Company’s functional currency.
Fair Value Information
In presenting our financial position, fair values are used for the measurement of various items in accordance with the applicable accounting standards. These fair values are based on market prices, where available, and are obtained from sources that are deemed to be reliable. Readers are cautioned that these values are subject to changes over time and are only valid at the balance sheet date. When quoted prices or observable market values do not exist, fair values are estimated using valuation models, which we believe are appropriate for their purpose. They require management to make significant assumptions with respect to future developments which are inherently uncertain and may therefore deviate from actual developments. Critical assumptions used are disclosed in the financial statements. In certain cases, independent valuations are obtained to support management’s determination of fair values.
Trademarks
We use various trademarks and tradenames, including without limitation “ProQR”, “Axiomer”, “Trident” and our corporate logo, that we use in connection with the operation of our business. Other trademarks, trade names or service marks of third parties referred to or incorporated by reference in this Annual Report are the property of their respective owners. Solely for convenience, the trademarks, trade names and service marks in this Annual Report may be referred to without the ®, ™ or SM symbols, but the omission of such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent permissible under applicable law, their rights thereto. We do not intend to use or display other companies’ trademarks, trade names and service marks to imply a relationship with, or endorsement or sponsorship of us, any other companies.
4
Cautionary Language Regarding Forward-looking Statements
This Annual Report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that involve substantial risks and uncertainties. The forward-looking statements are contained principally in Part I, Item 4.B “Business Overview,” Part I, Item 3.D. “Risk Factors,” and Part I, Item 5. “Operating and Financial Review and Prospects,” but are also contained elsewhere in this Annual Report. Forward-looking statements can be identified generally as those containing words as “aim,” “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “project,” “target,” “potential,” “will,” “would,” “could,” “should,” “continue,” or the negative of these terms, or other comparable terminology intended to identify statements about the future, although not all forward-looking statements contain these identifying words. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. The forward-looking statements contained in this Annual Report are based upon information available to us as of the date of this Annual Report and, while we believe we have a reasonable basis for each forward-looking statement contained this Annual Report, we caution you that these statements are based on a combination of facts and factors currently known by us and our expectations of the future, about which we cannot be certain.
By their nature, forward-looking statements involve risks and uncertainties because they relate to events, competitive dynamics and industry change, and depend on economic circumstances that may or may not occur in the future or may occur on longer or shorter timelines than anticipated. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Annual Report, we caution you that forward-looking statements are not guarantees of future performance and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. Known and unknown risks, uncertainties and other factors may cause our actual results, performance or achievements, including in relation to the research and (pre-)clinical development of our RNA editing platforms or any of our pipeline programs, to be materially different from our expectations. These forward-looking statements include, without limitation, statements about the following:
| ● | the cost, timing and results of preclinical studies and clinical trials and other development activities by us and our collaborative partners; |
| ● | the likelihood of our clinical programs being executed on timelines provided and reliance on our contract research organizations (“CROs”) and predictability of timely enrollment of subjects and patients to advance our clinical trials and maintain their own operations; |
| ● | our reliance on contract manufacturers to supply materials for research and development and the risk of supply interruption from a contract manufacturer; |
| ● | the potential for future data to alter initial and preliminary results of early-stage clinical trials; |
| ● | the unpredictability of the duration and results of the regulatory review of applications or clearances that are necessary to initiate and continue to advance and progress our clinical programs, and the ability to successfully submit the necessary applications or to obtain the necessary clearances; |
| ● | the ability to secure, maintain and realize the intended benefits of collaborations with partners; |
| ● | the possible impairment of, inability to obtain, and costs to obtain intellectual property rights; |
| ● | possible safety or efficacy concerns that could emerge as new data are generated in research and development; |
| ● | our ability to attract and retain key scientific and/or management personnel; our ability to obtain funding for our operations, including funding necessary to complete further development and commercialization of our product candidates, if approved; |
| ● | the accuracy of our estimates of our future revenue, expenses, capital requirements and needs for additional financing; |
| ● | our estimates regarding the market opportunities for our current and future programs and any future product candidates; |
5
| ● | the impact of global economic and political developments on our business, including rising inflation and capital market disruptions, economic sanctions and economic slowdowns or recessions that may result from such developments which could harm our research and development efforts as well as the value of our ordinary shares and our ability to access capital markets; |
| ● | the impact on our operations and activities that may be slowed or halted by shortage and/or pressure on supply and logistics on the global market and/or the impact of health epidemics, pandemics and other widespread outbreaks of contagious diseases, such as the COVID-19 pandemic or the post-COVID environment; |
| ● | general business, financial and accounting risks and risks related to litigation and disputes with third parties; and; |
| ● | the other risks and uncertainties, including those listed under the caption “Risk Factors.” |
In addition, even if our results, performance, financial condition and liquidity, and the development of the industry in which we operate are consistent with such forward-looking statements, they may not be predictive of results or developments in future periods. Among the factors that may result in differences are our expectations regarding risks and uncertainties related to the impact of the post-COVID environment on our business, geopolitical developments, operations, strategy, goals and anticipated milestones, including our ongoing and planned research activities, ability to conduct ongoing and planned clinical trials, clinical supply of current or future drug candidates, commercial supply of current or future approved products, and launching, marketing and selling current or future approved products, and our ability to obtain and maintain requisite regulatory approvals and to enroll patients in our planned clinical trials, additional interactions with regulatory authorities and expectations regarding future regulatory filings, our reliance on collaborations with third parties, estimating the commercial potential of our development programs and other risks indicated in the risk factors included in this report and other filings with the SEC.
Actual results could differ materially from our forward-looking statements due to a number of factors, including the risks set forth under the section “Risk Factors” and elsewhere in this Annual Report.
Any forward-looking statements that we make in this Annual Report are valid only as of the date of such statements, and we undertake no obligation to update such statements to reflect events or circumstances after the date of this Annual Report or to reflect the occurrence of unanticipated events.
This Annual Report contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. While we believe the market position, market opportunity and market size information included in this Annual Report is generally reliable, such information is inherently imprecise.
6
Part I
Item 1: Identity of Directors, Senior Management and Advisers
Not applicable.
Item 2: Offer Statistics and Expected Timetable
Not applicable.
Item 3: Key Information
A. [Reserved]
Not applicable.
B. Capitalization and Indebtedness
Not applicable.
C. Reasons for the Offer and Use of Proceeds
Not applicable.
D. Risk Factors
You should consider carefully the risks and uncertainties described below, together with all of the other information in this Annual Report, including the financial statements and the related notes included elsewhere in this Annual Report. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that adversely affect our business. If any of the following risks actually occurs, our business, financial condition, results of operations, and future prospects could be materially and adversely affected.
Risks Related to Our Capital Needs and Financial Position
We are a biopharmaceutical company with a history of losses. We expect to continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability, which could result in a decline in the market value of our ordinary shares.
We are a biopharmaceutical company with a limited operating history, engaged in the discovery and development of RNA-based therapeutics with our proprietary RNA editing platforms technology which could be used to develop new product candidates across multiple therapeutic areas. Since our inception in February 2012, we have devoted a significant portion of our resources to the development of our product candidates in inherited retinal disorders, including sepofarsen for Leber Congenital Amaurosis (“LCA”), ultevursen for Usher syndrome, and QR-1123 for autosomal dominant retinitis pigmentosa. Past resources have also been expended for the development of QR-504a for Fuchs endothelial corneal dystrophy, and eluforsen for cystic fibrosis, for which we have ceased making investments, and QR-313 for epidermolysis bullosa, which we have spun off. In August 2022 we announced our strategy to exclusively focus our resources on the development of our RNA editing platforms. As a result, we have ended the clinical development of sepofarsen and ultevursen, which we have sold to Laboratoires Théa S.A.S. (“Théa”) in December 2023. We have also ended the development of QR-1123 and QR-504a, for which we are currently seeking a partner to develop these programs. However, there can be no assurance that we will secure a partnership for the development of any of these product candidates. We have had significant operating losses since our inception. Our net losses for the years ended December 31, 2023, December 31, 2022 and December 31, 2021 were, € 27,735,000, € 64,204,000 and € 60,799,000, respectively. At December 31, 2023, we had an accumulated deficit of € 400,850,000. Substantially all of our losses have resulted from expenses incurred in connection with our research and development programs and from general and
7
administrative costs associated with our operations. Our RNA editing platforms are still in early stages of development and we are subject to the risks of failure inherent in the development of novel technologies and product candidates based on novel technologies.
To date, the only material income we have generated has been from the receipt of government research grants and collaboration agreements. Our ability to generate revenue from product sales depends upon our ability to successfully develop candidates with our RNA editing platforms, subsequently obtain regulatory approval for these candidates and successfully commercialize them and any other product candidates that we might develop, in-license or acquire in the future, enter into new collaboration agreements and generate milestone and royalty payments under existing or future collaboration agreements.
Even if we are able to successfully achieve regulatory approval for our product candidates, we do not know when any of these product candidates will generate revenue for us, if at all. We have not generated, and do not expect to generate, any product revenue for the foreseeable future, and we expect to continue to incur significant operating losses for the foreseeable future due to the cost of research and development of both RNA editing platforms and product candidates, preclinical studies and clinical trials and the regulatory approval process for product candidates. The amount of future losses is uncertain. Our ability to achieve profitability, if ever, will depend on, among other things, our ability to develop product candidates with our RNA editing platforms, us or any current or future collaborators successfully developing product candidates, obtaining regulatory approvals to market and commercialize product candidates, manufacturing any approved products on commercially reasonable terms, establishing a sales and marketing organization or suitable third party alternatives for any approved product and raising sufficient funds to finance business activities. If we or any current or future collaborators are unable to develop and commercialize one or more of our product candidates or if sales revenue from any product candidate that receives approval is insufficient, we will not achieve profitability, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
Our ability to generate revenue from our product candidates also depends on a number of additional factors, including our ability to:
| ● | successfully complete development activities, including the ongoing and planned preclinical and planned clinical studies for our product candidates; |
| ● | complete and submit New Drug Applications (“NDAs”) to the U.S. Food and Drug Administration (“FDA”), Marketing Authorization Applications (“MAAs”) to the European Medicines Agency (“EMA”), and comparable applications to other regulatory authorities and obtain regulatory approval for indications for which there is a commercial market; |
| ● | set a commercially viable price for any products for which we may receive approval; |
| ● | obtain commercial quantities of our products at acceptable cost levels; |
| ● | develop a commercial organization capable of sales, marketing and distribution for any product that we might intend to sell ourselves in the markets in which we have retained commercialization rights; |
| ● | find suitable partners to help us market, sell and distribute our approved products that we do not intend to sell ourselves in one or more markets; |
| ● | achieve acceptance among patients, clinicians and advocacy groups for any product we develop; and |
| ● | obtain coverage and adequate reimbursement for our products from third parties, including government payors. |
8
In addition, because of the numerous risks and uncertainties associated with product development, including the risk that our product candidates may not advance through development, as has been the case for product candidates for which we have suspended or ceased development activities, or be shown not to be safe and effective for their intended uses, the FDA, the EMA or other regulatory agencies may require additional clinical trials or preclinical studies or impose post-approval requirements for product candidates that do advance through development.
We are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. Even if we are able to complete the processes described above, we anticipate incurring significant costs associated with commercializing our product candidates.
On February 9, 2018, we entered into an agreement with Foundation Fighting Blindness (“FFB”), under which FFB has provided funding of $ 6.8 million to advance ultevursen into the clinic. Pursuant to the terms of the agreement, we were obligated to make certain repayments to FFB subject to development milestones. In December 2023, upon the occurrence of the sale of ultevursen to Théa, these payables were settled by means of a lump-sum payment in the amount of EUR 1.13 million and a percentage of earn-out payments for milestones and sales to be received by us from Théa, ranging from 5-10% Even if we are able to generate revenues from the sale of any of our product candidates or receive royalties from of our collaboration partners, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce our operations, which could result in a decline in the market value of our ordinary shares.
We will require additional capital to fund our operations and if we fail to obtain necessary financing, we will not be able to complete the development and commercialization of our product candidates.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts of cash to conduct further research and development, including with respect to our Axiomer and Trident platforms, and preclinical testing and clinical trials of our product candidates, to seek regulatory approvals for our product candidates and to launch and commercialize any product candidates for which we receive regulatory approval, including potentially building our own commercial organization to serve the United States, the European Union and certain other markets. As at December 31, 2023, we had €118,925,000 in cash and cash equivalents. Based on our current operating plans announced as part of our strategy update in August 2022, we believe that such existing cash and cash equivalents will be sufficient to fund our anticipated level of operations into mid-2026. However, our future capital requirements and the period for which our existing resources will support our operations may vary significantly from what we expect. Our monthly spending levels will vary based on new and ongoing development and corporate activities. Because the length of time and activities associated with successful development of our RNA editing platforms and product candidates is highly uncertain, we are unable to estimate the actual funds we will require for development and any approved marketing and commercialization activities relating to product candidates. Further, changing circumstances, some of which may be beyond our control, could cause us to consume capital significantly faster than we currently anticipate, and we may need to seek additional funds sooner than planned. Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
| ● | the preclinical and clinical development plans we establish for our product candidates; |
| ● | the number and characteristics of product candidates that we develop or may in-license; |
| ● | the extent to which we in-license or acquire rights to other products, product candidates or technologies; |
| ● | the number and characteristics of programs that we may pursue in our innovation unit, including the development of RNA editing platforms; |
| ● | the achievement of milestones that trigger payments under our existing or future collaboration agreements; |
| ● | our ability to establish collaboration arrangements for the development of our product candidates on favorable terms, if at all; |
9
| ● | the outcome, timing and cost of meeting regulatory requirements established by the FDA, the EMA and other comparable foreign regulatory authorities; |
| ● | the cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual property rights; |
| ● | the cost of defending any intellectual property disputes, including patent infringement actions brought by third parties against us or our product candidates; |
| ● | the effect of competing technological and market developments; |
| ● | the cost and timing of completion of commercial-scale outsourced manufacturing activities; and |
| ● | the cost of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our own. |
We cannot be certain that additional funding will be available on acceptable terms, or at all. For instance, the trading prices for our ordinary shares and for other biopharmaceutical companies have been highly volatile. As a result, we may face difficulties raising capital through sales of our equity or debt securities or such sales may be on unfavorable terms. Similarly, adverse market or macroeconomic conditions or market volatility resulting from global economic developments, political unrest, high inflation, rising interest rates, the post-COVID environment, future public health epidemics or other factors, could materially and adversely affect our ability to consummate an equity or debt financing on favorable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our products or product candidates or one or more of our other research and development initiatives such as our RNA editing platforms. In order to raise additional capital, we may seek a combination of private and public equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, existing ownership interests may be diluted and the terms of such financings may include liquidation or other preferences that adversely affect the rights of existing shareholders. We also could be required to seek collaborators for one or more of our current or future product candidates at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be acceptable or relinquish or license on unfavorable terms our rights to technologies or product candidates that we otherwise would seek to develop or commercialize ourselves.
While we were founded in 2012, we announced our plans to refocus our business on our RNA editing platforms in 2022, and it may be difficult to assess the future viability of our business and our strategy.
We were founded in February 2012 and began operations in May 2012, since which time we have primarily focused our efforts on development activities for our product candidates, including acquiring and developing product and technology rights and entering into collaborations. We have revised our strategy from time to time, including most recently in August 2022 when we announced our plan to focus exclusively on our RNA editing platforms, therewith withdrawing from the ophthalmology space. Consequently, any predictions about our future success, performance or viability may not be as accurate as they could be if we had a longer operating history, more experience with research and development initiatives and clinical development or approved products on the market.
10
We or third parties upon whom we depend may be adversely affected by natural disasters, geopolitical developments and/or global health pandemics, and our business, financial condition and results of operations could be adversely affected.
The occurrence of unforeseen or catastrophic events, including extreme weather events and other natural disasters, man-made disasters, or the emergence of epidemics or pandemics, or geopolitical events, depending on their scale, may cause different degrees of damage to the national and local economies, such as recessions, rising interest rates, inflation, fuel prices fluctuations, foreign currency fluctuations, international tariffs, boycotts, curtailment of trade and other business restrictions, and could cause a disruption in our operations and have a material adverse effect on our financial condition and results of operations. Man-made disasters, pandemics, and other events connected with the regions in which we operate could have similar effects. If a natural disaster, health pandemic, or other event beyond our control occurred that prevented us from using all or a significant portion of our office and/or lab spaces, damaged critical infrastructure, such as our manufacturing facilities or our manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations or supplies, it may be difficult for us to continue our business for a substantial period of time.
The effects of health epidemics, pandemics and other widespread outbreaks of contagious disease, such as the COVID-19 pandemic and the post-COVID environment, may materially and adversely affect our business and our financial results.
Significant outbreaks of contagious diseases and other adverse public health developments could have a material impact on our business operations and operating results. For instance, the COVID-19 pandemic and the post-COVID environment, including supply chain, labor market and other disruptions, as well as volatility in the global financial markets, in each case, driven by the pandemic, have affected and may further negatively impact segments of the global economy and our operations or the operations of our vendors, suppliers and business partners, including the third-party contract research organizations (“CROs”), clinical sites and other vendors that we rely upon to carry out our preclinical studies or planned clinical trials of our product candidates or the operations of our third-party manufacturers and other suppliers, which could result in delays or disruptions in the supply of our product candidates. The negative impact COVID-19 or the post-COVID environment has on patient enrollment, site staffing or treatment or the timing and execution of our clinical trials and preclinical studies has caused and could cause further delays to our clinical trial activities, which could adversely affect our ability to obtain regulatory approval for and to commercialize our product candidates, increase our operating expenses and have a material adverse effect on our business and financial results. COVID-19 and the post-COVID environment have also caused volatility in the global financial markets, including inflationary headwinds, which may negatively affect our ability to raise additional capital on attractive terms or at all.
We cannot presently predict the scope and severity of any potential business shutdowns or disruptions, but if we or any of the third parties with whom we engage were to experience prolonged business shutdowns or other disruptions, our ability to conduct our business in the manner and on the timelines presently planned could be materially and negatively affected, which could have a material adverse impact on our business, results of operation and financial condition.
Increased attention to, and evolving expectations for, environmental, social, and governance (“ESG”) initiatives could increase our costs, harm our reputation, or otherwise adversely impact our business.
Companies across industries are facing increasing scrutiny from a variety of stakeholders related to their ESG and sustainability practices, including practices associated with climate change. Expectations regarding voluntary ESG initiatives and disclosures may result in increased costs (including but not limited to increased costs related to compliance, stakeholder engagement, contracting and insurance), enhanced compliance or disclosure obligations, or other adverse impacts to our business, financial condition, or results of operations.
While we may at times engage in voluntary initiatives (such as voluntary disclosures, certifications, or goals, among others) to improve the ESG profile of the Company, such initiatives may be costly and may not have the desired effect. Moreover, we may not be able to successfully complete such initiatives due to factors that are within or outside of our control. Even if this is not the case, our actions may subsequently be determined to be insufficient by various stakeholders, and we may be subject to investor or regulator engagement on our ESG efforts, even if such initiatives are currently voluntary.
11
Certain market participants, including major institutional investors and capital providers, use third-party benchmarks and scores to assess companies’ ESG profiles in making investment or voting decisions. Unfavorable ESG ratings could lead to increased negative investor sentiment towards us, which could negatively impact our share price as well as our access to and cost of capital. To the extent ESG matters negatively impact our reputation, it may also impede our ability to compete as effectively to attract and retain employees, which may adversely impact our operations.
In addition, we expect there will likely be increasing levels of regulation, disclosure-related and otherwise, with respect to ESG matters. For example, the SEC has published propose rules that would require companies to provide significantly expanded climate-related disclosures in their periodic reporting, which may require us to incur significant additional costs to comply, including the implementation of significant additional internal controls processes and procedures regarding matters that have not been subject to such controls in the past, and impose increased oversight obligations on our management and board of directors. These and other changes in stakeholder expectations will likely lead to increased costs as well as scrutiny that could heighten all of the risks identified in this risk factor. Additionally, our business partners may be subject to similar expectations, which may augment or create additional risks, including risks that may not be known to us.
Risks Related to the Development and Regulatory Approval of our Product Candidates
Our business depends in part on the success of our product candidates. We cannot be certain that we will be able to successfully complete the clinical development of, obtain regulatory approval for, or successfully commercialize our product candidates.
Our business depends in part on our ability to develop, secure regulatory approval for and commercialize one or more product candidates, including any product candidates resulting from our initial pipeline programs using our Axiomer technology, including AX-0810 for Cholestatic Diseases targeting NTCP and AX-1412 for Cardiovascular Disease (“CVDs”) targeting B4GALT1. However, the development of drug candidates is lengthy, uncertain and expensive, and there can be no assurance that we will achieve such goals. For example, in the past, we have focused our efforts on the development of product candidates in our ophthalmology portfolio and, previously, for cystic fibrosis, none of which we are planning to continue developing at this time. In February 2022, we announced that top-line results from our Phase 2/3 pivotal trial of sepofarsen did not meet its primary endpoint.
In addition, as part of our strategy update in April 2022, we suspended the early phase clinical trials for our QR-1123 and QR-504a programs and suspended all other research activities related to our inherited retinal disease pipeline. In August 2022, we announced our strategy to shift our focus and resources exclusively to the development of our RNA editing platforms, ending the clinical development of sepofarsen and ultevursen and ultimately ending all activity related to the broader ophthalmology portfolio. In December 2023, we have sold the assets sepofarsen and ultevursen to Théa, which Théa will further develop.
The clinical trials and manufacturing and marketing of our future product candidates will be subject to extensive and rigorous review and regulation by numerous government authorities in the United States, the European Union and other jurisdictions where we intend to test and, if approved, market our product candidates. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through preclinical testing and clinical trials that the product candidate is safe and effective for use in each target indication, and potentially in specific patient populations, including the pediatric population. This process can take many years and may include post-marketing studies and surveillance, which would require the expenditure of substantial resources beyond our existing funds. Of the large number of drugs in development for approval in the United States and the European Union, only a small percentage successfully complete the FDA or EMA regulatory approval processes, as applicable, and are commercialized. Accordingly, even if we are able to obtain the requisite financing to continue to fund our research, development and clinical programs, we cannot be certain that any of our product candidates will be successfully developed or commercialized.
12
In addition, if Théa or a third party collaborator resumes or were to resume the advancement of sepofarsen, ultevursen, QR-1123 or QR-504a, or commence new clinical programs of our product candidates in the future, the successful clinical development, regulatory approval and commercialization of any of these product candidates would require additional or new preclinical and clinical testing and substantial additional or new clinical development and regulatory approval efforts before we are permitted to commence their commercialization, if ever. It would be several years before a pivotal trial could be completed for any of such other product candidates, if ever.
We may not be able to file INDs or IND amendments or similar applications to commence clinical trials of our product candidates on the timelines we expect, and even if we are able to, the FDA or similar foreign regulatory authority may not permit us to proceed.
We may not be able to file INDs or similar applications for our product candidates on the timelines we expect. For example, we may experience manufacturing delays or other delays with IND-enabling studies. Moreover, we cannot be sure that submission of an IND or similar application will result in the FDA or similar foreign regulatory authority allowing clinical trials to begin, or that, once begun, issues will not arise that result in the suspension or termination of clinical trials. Additionally, even if such regulatory authorities agree with the design and implementation of the clinical trials set forth in an IND or similar application, we cannot guarantee that such regulatory authorities will not change their requirements in the future. These considerations apply to new clinical trials we may submit as amendments to existing INDs or to a new IND or similar application. Any failure to file INDs or similar applications on the timelines we expect or to obtain authorization to begin our trials may prevent us from completing our clinical trials or commercializing our products on a timely basis, if at all.
The regulatory approval processes of the FDA, the EMA and comparable foreign regulatory authorities are lengthy, time-consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.
We are not permitted to market our product candidates in the United States or the European Union until we receive an NDA approval from the FDA or an MAA approval from the European Commission, respectively, or in any other foreign countries until we receive the requisite approval from such countries. Prior to submitting an NDA to the FDA or an MAA to the EMA for approval of any product candidate, we will need to complete preclinical and toxicology studies, as well as proof-of-concept studies and Phase 1, Phase 2 and Phase 3 clinical trials for such candidate. While we intend to submit marketing applications for our product candidates that successfully complete clinical development, there can be no assurance that we will be able to do so in a timely manner or at all. Successfully initiating and completing clinical programs and obtaining approval of an NDA or a MAA is a complex, lengthy, expensive and uncertain process, and the FDA, the EMA or other comparable foreign regulatory authorities may delay, limit or deny approval of our product candidates for many reasons, including, among others:
| ● | we may not be able to demonstrate that our product candidates are safe and effective in treating patients to the satisfaction of the FDA or the EMA; |
| ● | the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA or the EMA for marketing approval; |
| ● | the FDA or the EMA may disagree with the number, design, size, conduct or implementation of our clinical trials; |
| ● | the FDA or the EMA may require that we conduct additional clinical trials, including pivotal trials; |
| ● | the FDA or the EMA or other applicable foreign regulatory agencies may not approve the formulation, labeling or specifications of our product candidates; |
| ● | the CROs that we retain to conduct our clinical trials may take actions outside of our control that materially adversely impact our clinical trials; |
13
| ● | the FDA or the EMA may find the data from preclinical studies and clinical trials insufficient to demonstrate that the clinical and other benefits of our products outweigh their safety risks; |
| ● | the FDA or the EMA may disagree with our interpretation of data from our preclinical studies and clinical trials; |
| ● | the FDA or the EMA may not accept data generated at our clinical trial sites; |
| ● | if our NDAs or MAAs, if and when submitted, are reviewed by the FDA or the EMA, as applicable, these regulatory agencies may have difficulties scheduling the necessary review meetings in a timely manner, may recommend against approval of our application or may require, as a condition of approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
| ● | the FDA may require development of a Risk Evaluation and Mitigation Strategy as a condition of approval or post-approval, and the EMA may grant only conditional approval or impose specific obligations as a condition for marketing authorization, or may require us to conduct post-authorization safety studies; |
| ● | the FDA, the EMA or other applicable foreign regulatory agencies may not approve the manufacturing processes or facilities of third-party manufacturers with which we contract; or |
| ● | the FDA or the EMA may change their approval policies or adopt new regulations. |
Any of these factors, many of which are beyond our control, could jeopardize our ability to obtain regulatory approval for and successfully market our product candidates. Any such setback in our pursuit of regulatory approval would have a material adverse effect on our business and prospects.
We may never realize a return on our investment of resources and cash from our drug discovery collaborations.
In addition to developing our own pipeline of product candidates, we also conduct drug discovery activities for or with collaborators who are also engaged in drug discovery and development, which could include pre-commercial biotechnology companies and large pharmaceutical companies. Under these collaborations, we might provide the benefit of our drug discovery platforms and platform experts who identify molecules that have activity against one or more specified targets, among other resources. In consideration, we have received, and might receive in the future, (i) equity investments; (ii) upfront fees; and/or (iii) the right to receive option fees, cash milestone payments upon the achievement of specified development, regulatory, or commercial sales milestones for the drug discovery targets, and potential royalties. Our ability to receive fees and payments and realize returns from our drug discovery collaborations in a timely manner, or at all, is subject to a number of risks, including but not limited to the following:
| ● | our collaborators may incur unanticipated costs or experience delays in completing, or may be unable to complete, the development and commercialization of any drug candidates; |
| ● | collaborators have significant discretion in determining the amount and timing of efforts and resources that they will apply to our collaborations and may not perform their obligations as expected; |
| ● | collaborators may decide not to pursue development or commercialization of drug candidates for various reasons, including results of clinical trials or other studies, changes in the collaborator’s strategic focus or available funding, their desire to develop products that compete directly or indirectly with our drug candidates, or external factors (such as an acquisition or industry slowdown) that divert resources or create competing priorities; |
| ● | existing collaborators and potential future collaborators may begin to perceive us to be a competitor more generally, particularly as we advance our internal drug discovery programs, and therefore may be unwilling to continue existing collaborations, or enter into new collaborations, with us; |
14
| ● | a collaborator may fail to comply with applicable regulatory requirements regarding the development, manufacture, distribution, or marketing of a drug candidate or product; |
| ● | a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products; |
| ● | disagreements with collaborators, including disagreements over intellectual property or proprietary rights, contract interpretation, or the preferred course of development, might cause delays or terminations of the research, development, or commercialization of drug candidates, or might result in litigation or arbitration; |
| ● | collaborators may not properly obtain, maintain, enforce, defend, or protect our or their intellectual property or proprietary rights, or they may use our proprietary information in such a way as to potentially lead to disputes or legal proceedings that could jeopardize or invalidate our or their intellectual property or proprietary rights; |
| ● | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates; |
| ● | collaborators may infringe, misappropriate, or otherwise violate the intellectual property or proprietary rights of third parties, which may expose us to litigation and potential liability; and |
| ● | drug discovery collaborations may be terminated prior to our receipt of any significant value. |
Failures or delays in the commencement or completion of our preclinical studies or planned or future clinical trials of our future product candidates could result in increased costs to us and could delay, prevent or limit our ability to generate revenue and continue our business.
Successful completion of preclinical studies and clinical trials is a prerequisite to submitting an NDA to the FDA or an MAA to the EMA and, consequently, the ultimate approval and commercial marketing of our product candidates. Clinical trials are expensive, difficult to design and implement, can take many years to complete and are uncertain as to outcome. A product candidate can unexpectedly fail at any stage of clinical development. The historical failure rate for product candidates is high due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables. We do not know whether our future clinical trials will begin or be completed on schedule, if at all, as the commencement and completion of clinical trials can be delayed or prevented for a number of reasons, including, among others:
| ● | delays in reaching or failing to reach agreement on acceptable terms with prospective CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | inadequate quantity or quality of a product candidate or other materials necessary to conduct clinical trials; |
| ● | difficulties obtaining Institutional Review Board (“IRB”) or ethics committee approval to conduct a clinical trial at a prospective site or sites, or institutional biosafety committee (“IBC”) approval, if applicable; |
| ● | challenges in recruiting and enrolling patients to participate in clinical trials, including the size and nature of the patient population, the proximity of patients to clinical sites, eligibility criteria for the clinical trial, the nature of the clinical trial protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications; |
| ● | severe or unexpected drug-related side effects experienced by patients in our clinical trials or by individuals using drugs similar to our product candidates; |
| ● | reports from preclinical or clinical testing of other RNA therapies that raise safety or efficacy concerns; |
15
| ● | difficulties retaining patients who have enrolled in a clinical trial but may be prone to withdraw due to lack of efficacy, side effects, personal reasons or loss of interest; or |
| ● | force majeure events, such as the COVID-19 pandemic and the post-COVID environment. |
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, the EMA, the IRBs at the sites where the IRBs are overseeing a clinical trial, upon recommendation of a data safety monitoring board (“DSMB”) overseeing the clinical trial at issue, or by other regulatory authorities due to a number of factors, including, among others:
| ● | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| ● | inspection of the clinical trial operations or trial sites by the FDA, the EMA or other regulatory authorities that reveals deficiencies or violations that require us to undertake corrective action, including the imposition of a clinical hold; |
| ● | unforeseen safety issues, including any that could be identified in our ongoing toxicology studies, adverse side effects or lack of effectiveness; |
| ● | changes in government regulations or administrative actions; |
| ● | problems with clinical supply materials; and |
| ● | lack of adequate funding to continue the clinical trial. |
Positive results from early-stage clinical trials and preclinical testing of our future product candidates are not necessarily predictive of the results of later clinical trials for these product candidates. If we cannot achieve positive results in our later stage clinical trials for our product candidates, we may be unable to successfully develop, obtain regulatory approval for and commercialize them.
We may from time to time publish results from preclinical studies and clinical trials of our product candidates. Positive results from early-stage clinical trials and preclinical testing of our product candidates in vitro and in vivo may not necessarily be predictive of the results from later-stage clinical trials. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in later-stage clinical trials after achieving positive results in preclinical and early-stage clinical trials, and we cannot be certain that we will not face similar setbacks. These setbacks have been caused by, among other things, preclinical findings made while clinical trials were already underway or safety or efficacy observations made in clinical trials, including adverse events. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA or EMA approval. If we fail to produce positive results in our clinical trials for any product candidates, the development timeline and regulatory approval and commercialization prospects for those product candidates, and, correspondingly, our business and financial prospects would be materially adversely affected.
16
Additionally, some of our clinical trials have utilized and may in the future utilize an “open-label” trial design. An open-label clinical trial is one where both the patient and investigator know whether the patient is receiving the investigational product candidate or either an existing approved drug or placebo. Most typically, open-label clinical trials test only the investigational product candidate and sometimes may do so at different dose levels. Open-label clinical trials are subject to various limitations that may exaggerate any therapeutic effect as patients in open-label clinical trials are aware when they are receiving treatment. Open-label clinical trials may be subject to a patient bias where patients perceive their symptoms to have improved merely due to their awareness of receiving an experimental treatment. Moreover, patients selected for early clinical studies often include the most severe sufferers and their symptoms may have been bound to improve notwithstanding the new treatment. In addition, open-label clinical trials may be subject to an investigator bias where those assessing and reviewing the physiological outcomes of the clinical trials are aware of which patients have received treatment and may interpret the information of the treated group more favorably given this knowledge. The results from an open-label trial may not be predictive of future clinical trial results with any of our product candidates when studied in a controlled environment with a placebo or active control.
Changes in regulatory requirements or guidance from the FDA, EMA or other applicable regulatory authorities, or unanticipated events during our preclinical or clinical development activities may occur, which may result in changes to requirements, which could result in increased costs to us and could delay our development timeline.
Changes in regulatory requirements, or guidance from the FDA, EMA, or other applicable regulatory authorities, imposition of additional preclinical or clinical study requirements by applicable regulatory authorities or unanticipated events during our preclinical or clinical development may force us to amend study protocols or conduct additional studies or trials or involve delays to our preclinical and clinical studies and overall development strategy, all of which could result in increased costs for the development of our product candidates. We may experience delays if we are required by the FDA, EMA, or other applicable regulatory authorities, or if we voluntarily decide to, redesign or restructure a clinical trial once initiated. Amendments to our clinical trial protocols may require resubmission to the applicable regulatory authorities and IRBs or ethics committees for review and approval, which may adversely impact the cost, timing and/or successful completion of a clinical trial. We may be delayed in our clinical development if we are unable to obtain alignment with the FDA, EMA, or other applicable regulatory authorities on an acceptable clinical trial design or statistical analysis plan for any of our clinical trials. Additionally, the FDA, EMA, or other applicable regulatory authorities may require us to conduct additional clinical trials, including pivotal trials, to support a marketing application. If we experience delays completing, or if we terminate, any of our clinical trials, or if we are required to conduct additional clinical trials, the commercial prospects for our product candidates may be harmed and our ability to generate product revenue will be delayed.
17
Our RNA technologies are unproven in the disease indications for which they are being developed and tested and may not lead to the development of, or result in, marketable products.
We are developing RNA editing platforms and a pipeline of product candidates using these proprietary RNA technologies for genetic disorders with unmet need. We believe that targeting the mRNA to restore the production of functional protein is a unique approach that offers advantages versus small molecule, gene therapy and other therapeutic approaches. However, the scientific research that forms the basis of our efforts to develop product candidates is both preliminary and limited. The mechanism of action of our compounds could be different from what we today hypothesize. Also, we may discover that the molecules that we develop do not possess certain properties required for a drug to be effective, such as the ability to remain stable in the human body for the period of time required for the drug to reach the target tissue or the ability to cross the cell wall and enter into cells within the target tissue for effective delivery. We may spend substantial funds attempting to introduce these properties and may never succeed in doing so. For example, while we have discovered and are developing our novel RNA editing platforms and will focus our resources exclusively on these RNA editing platforms as announced as part of our strategy update in August 2022, there can be no assurance that we will be able to leverage our technology to create viable product candidates to advance into the clinic, or develop those candidates to submit for regulatory approval. In addition, product candidates based on RNA technologies may demonstrate different chemical and pharmacological properties in humans than they do in laboratory studies or animals. Even if our product candidates have successful results in animal studies, they may not demonstrate the same chemical and pharmacological properties in humans and may interact with human biological systems or other existing treatments in unforeseen, ineffective or harmful ways. Our RNA technologies may provoke an unwanted immune response, or immunogenicity, which may neutralize the therapeutic effects of our product candidates and could result in harmful outcomes for patients. As a result, we may never succeed in developing a marketable product, and in turn we may not become profitable and the value of our ordinary shares would decline.
While there are a number of approved therapies based on the oligonucleotide modality, there are no approved therapies based on the editing technology underlying our novel editing mechanism which uses Adenosine Deaminase Acting on RNA (“ADAR”). This may increase the complexity, uncertainty and length of the regulatory approval process for our product candidates. Neither we nor any future collaborator may receive approval to market and commercialize any product candidate. Even if we or a collaborator were to obtain regulatory approval, the approval may be for disease indications or patient populations that are not as broad as we intended or may require labeling that includes significant use or distribution restrictions or safety warnings. We or a collaborator may be required to perform additional or unanticipated clinical trials to obtain approval or be subject to post-marketing testing requirements to maintain regulatory approval. If our RNA technology proves to be ineffective, unsafe or commercially unviable, our entire platforms and pipeline would have little, if any, value, which would have a material adverse effect on our business, financial condition, results of operations and prospects.
Failure to obtain regulatory approval in jurisdictions outside the United States and the European Union would prevent our product candidates from being marketed in those jurisdictions.
In order to market and sell our products in jurisdictions other than the United States and the European Union, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The regulatory approval process outside the United States and the European Union generally includes all of the risks associated with obtaining FDA and EMA approval, but can involve additional testing. We may need to partner with third parties in order to obtain approvals outside the United States and the European Union. In addition, in many countries worldwide, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We may not obtain approvals from regulatory authorities outside the United States and the European Union on a timely basis, if at all. Even if we were to receive approval in the United States or the European Union, approval by the FDA or the EMA does not ensure approval by regulatory authorities in other countries or jurisdictions. Similarly, approval by one regulatory authority outside the United States and the European Union would not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA or the EMA. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market. If we are unable to obtain approval of our product candidates by regulatory authorities in other foreign jurisdictions, the commercial prospects of those product candidates may be significantly diminished, and our business prospects could decline.
18
A breakthrough therapy designation by the FDA for our product candidates may not lead to a faster development or regulatory review or approval process, and it does not increase the likelihood that our product candidates will receive marketing approval.
We may seek a breakthrough therapy designation for our product candidates in the future. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapies on one or more clinically significant endpoints. For product candidates that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Product candidates designated as breakthrough therapies by the FDA may also be eligible for accelerated approval.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe that any of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. While the FDA has released guidance as to the criteria it uses in designating drugs as breakthrough therapies, we cannot be sure that our product candidates will meet the FDA’s qualifying criteria for such designation. In any event, the receipt of a breakthrough therapy designation for a product candidate may not result in a faster development process, review or approval compared to drugs considered for approval under conventional FDA procedures and also does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that the products no longer meet the conditions for such qualification or decide that the time period for FDA review or approval will not be shortened.
We may seek designation for our Axiomer and Trident RNA editing platforms as designated platform technologies, but we might not receive such designation, and even if we do, such designation may not lead to a faster development, regulatory review or approval process.
We may seek designation for our Axiomer and Trident RNA editing platforms as designated platform technologies. Under the Food and Drug Omnibus Reform Act of 2022 (“FDORA”), a platform technology incorporated within or utilized by a drug or biological product is eligible for designation as a designated platform technology if (1) the platform technology is incorporated in, or utilized by, a drug approved under an NDA or BLA; (2) preliminary evidence submitted by the sponsor of the approved or licensed drug, or a sponsor that has been granted a right of reference to data submitted in the application for such drug, demonstrates that the platform technology has the potential to be incorporated in, or utilized by, more than one drug without an adverse effect on quality, manufacturing, or safety; and (3) data or information submitted by the applicable person indicates that incorporation or utilization of the platform technology has a reasonable likelihood to bring significant efficiencies to the drug development or manufacturing process and to the review process. A sponsor may request the FDA to designate a platform technology as a designated platform technology concurrently with, or at any time after, submission of an IND for a drug that incorporates or utilizes the platform technology that is the subject of the request. If so designated, the FDA may expedite the development and review of any subsequent original NDA or BLA for a drug that uses or incorporates the platform technology. Even if we believe our Axiomer and Trident platforms meet the criteria for such designation, the FDA may disagree and instead determine not to grant such designation. In addition, the receipt of such designation for a platform technology does not ensure that a drug will be developed more quickly, be reviewed by the FDA faster, or receive FDA approval. Moreover, the FDA may revoke a designation if the FDA determines that a designated platform technology no longer meets the criteria for such designation.
19
Our therapeutic candidates are based on a novel mechanism of action, which makes it difficult to predict the time and cost of development and of subsequently obtaining regulatory approval, if at all.
Although we have discovered and are developing our novel RNA editing platforms and will focus our resources exclusively on these RNA editing platforms, as announced as part of our strategy update in August 2022, there can be no assurance that we will be able to leverage our technology to create viable product candidates to advance into the clinic, or develop those candidates to submit for regulatory approval. In addition, product candidates based on RNA technologies may demonstrate different chemical and pharmacological properties in humans than they do in laboratory studies or animals. Even if our product candidates have successful results in animal studies, they may not demonstrate the same chemical and pharmacological properties in humans and may interact with human biological systems or other existing treatments in unforeseen, ineffective or harmful ways. Our RNA technologies may provoke an unwanted immune response, or immunogenicity, which may neutralize the therapeutic effects of our product candidates and could result in harmful outcomes for patients. As a result, we or our partners may never succeed in developing a marketable product based on our platforms, and in turn we may not become profitable and the value of our ordinary shares would decline.
Furthermore, the FDA and the EMA have relatively limited experience with therapeutics based on RNA. This may increase the complexity, uncertainty and length of the regulatory approval process for our product candidates. Neither we nor any future collaborator may receive approval to market and commercialize any product candidate. Even if we or a collaborator were to obtain regulatory approval, the approval may be for disease indications or patient populations that are not as broad as we intended or may require labeling that includes significant use or distribution restrictions or safety warnings. We, or a collaborator, may be required to perform additional or unanticipated clinical trials to obtain approval or be subject to post-marketing testing requirements to maintain regulatory approval. If our RNA technology proves to be ineffective, unsafe or commercially unviable, our entire platforms and pipeline would have little, if any, value, which would have a material adverse effect on our business, financial condition, results of operations and prospects.
Risks Related to Our Dependence on Third Parties
If a collaborative partner terminates or fails to perform its obligations under an agreement with us, the commercialization of our product candidates, if approved, could be delayed or terminated.
We are party to, and we may from time to time in the future pursue, collaborative arrangements for the development and commercialization of product candidates, if approved. We have executed and continue to consider strategic alternatives that include spinouts, out-licensing or collaborative partnerships to develop and commercialize our RNA technologies, including our Axiomer program, or programs. For example, in 2021, we entered into a licensing and research collaboration agreement with Eli Lilly and Company (“Lilly”), as amended in 2022, which expanded our collaboration, focused on the discovery, development, and commercialization of potential new medicines primarily in the peripheral nervous system and central nervous system and metabolic diseases. If any of our collaborative partners in future collaborative arrangements for the commercialization of product candidates or similar arrangements does not devote sufficient time and resources to the collaboration arrangement with us, we may not realize the potential commercial benefits of the arrangement, and our results of operations may be materially adversely affected. In addition, if any such collaboration partner were to breach or terminate its arrangements with us, the commercialization of our product candidates could be delayed, curtailed or terminated.
Much of the potential revenue from current and future collaborations may consist of contingent payments, such as payments for achieving regulatory milestones or royalties payable on sales of drugs. The milestone and royalty revenue that we may receive under these collaborations will depend upon our collaborators’ ability to successfully develop, introduce, market and sell new products. In addition, collaborators may decide to enter into arrangements with third parties to commercialize products developed under collaborations using our technologies, which could reduce the milestone and royalty revenue that we may receive, if any. Future collaboration partners may fail to develop or effectively commercialize products using our products or technologies because they:
20
| ● | decide not to devote the necessary resources due to internal constraints, such as limited personnel with the requisite expertise, limited cash resources or specialized equipment limitations, or the belief that other drug development programs may have a higher likelihood of obtaining marketing approval or may potentially generate a greater return on investment; |
| ● | decide to pursue other technologies or develop other product candidates, either on their own or in collaboration with others, including our competitors, to treat the same diseases targeted by our own collaborative programs; |
| ● | do not have sufficient resources necessary to carry the product candidate through clinical development, marketing approval and commercialization; or |
| ● | cannot obtain the necessary marketing approvals. |
Moreover, competition may negatively impact a partner’s focus on, and commitment to, our product candidates and, as a result, could delay or otherwise negatively affect the commercialization of such product candidate. The failure of any current or future collaboration partners to develop or effectively commercialize our product candidates, if approved, for any of these reasons would have a material adverse effect on our operating results and financial condition.
For sepofarsen and ultevursen, we will depend on Théa to develop and conduct clinical trials with, obtain regulatory approvals for, and if approved, market and sell these product candidates. We may also pursue similar relationships with other development and commercialization collaborators for additional product candidates in the future or expand the reach with current collaborators. If such collaborators fail to perform as expected, the potential for us to generate future revenue from such product candidates would be significantly reduced and our business would be harmed.
For certain product candidates, we depend, or will depend, on our development and commercial collaborators to develop, conduct clinical trials of, and, if approved, commercialize product candidates.
Our current collaborations and any future collaborations that we enter into are subject to numerous risks, including:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to the collaborations; |
| ● | collaborators may not perform their obligations as expected or fail to fulfill their responsibilities in a timely manner, or at all; |
| ● | collaborators may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue or renew development or commercialization programs based on preclinical studies or clinical trial results, changes in the collaborators’ strategic focus or available funding or external factors, such as an acquisition, that divert resources or create competing priorities; |
| ● | collaborators may delay preclinical studies or clinical trials, provide insufficient funding for clinical trials, stop a preclinical study or clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
| ● | we may not have access to, or may be restricted from disclosing, certain information regarding product candidates being developed or commercialized under a collaboration and, consequently, may have limited ability to inform our shareholders about the status of such product candidates; |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
21
| ● | the collaborations may not result in product candidates to develop and/or preclinical studies or clinical trials conducted as part of the collaborations may not be successful; |
| ● | product candidates developed with collaborators may be viewed by our collaborators as competitive with their own product candidates or products, which may cause collaborators to stop commercialization of our product candidates; |
| ● | a collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may not commit sufficient resources to the marketing and distribution of any such product candidate; and |
| ● | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation. |
In addition, certain collaboration and commercialization agreements provide our collaborators with rights to terminate such agreements, which rights may or may not be subject to conditions, and which rights, if exercised, would adversely affect our product development efforts and could make it difficult for us to attract new collaborators. In that event, we would likely be required to limit the size and scope of efforts for the development and commercialization of such product candidates or products; we would likely be required to seek additional financing to fund further development or identify alternative strategic collaborations; our potential to generate future revenue from royalties and milestone payments from such product candidates or products would be significantly reduced, delayed or eliminated; and it could have an adverse effect on our business and future growth prospects. Our rights to recover tangible and intangible assets and intellectual property rights needed to advance a product candidate or product after termination of a collaboration may be limited by contract, and we may not be able to advance a program post-termination.
We rely on third-party manufacturers and suppliers and we intend to rely on third parties to produce preclinical, clinical and commercial supplies of our product candidates.
We rely on third parties to supply the materials and components for, and manufacture, our research and development, preclinical and clinical trial supplies. We do not own manufacturing facilities or supply sources for such components and materials. There can be no assurance that our supply of research and development, preclinical and clinical development drugs and other materials will not be limited, interrupted, restricted in certain geographic regions or of satisfactory quality or continue to be available at acceptable prices. In particular, any replacement of our drug product formulation manufacturer could require significant effort and expertise because there may be a limited number of qualified replacements.
The manufacturing process for a product candidate is subject to FDA, EMA and other foreign regulatory authority review. Suppliers and manufacturers must meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with regulatory standards such as cGMP. In the event that any of our suppliers or manufacturers fails to comply with such requirements or fails to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the materials ourselves, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on reasonable terms, if at all. In some cases, the technical skills or technology required to manufacture our product candidates may be unique or proprietary to the original manufacturer and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills or technology to another third party and a feasible alternative may not exist. These factors would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture our product candidates. If we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturer or manufacturing process will produce our product candidate according to the specifications previously submitted to the FDA, the EMA or another regulatory authority. The delays associated with the verification of a new manufacturer or manufacturing process could negatively affect our ability to develop product candidates in a timely manner or within budget.
22
We expect to continue to rely on third party manufacturers if we receive regulatory approval for any product candidate. To the extent that we have existing, or enter into future, manufacturing arrangements with third parties, we will depend on these third parties to perform their obligations in a timely manner consistent with contractual and regulatory requirements, including those related to quality control and assurance. If we are unable to obtain or maintain third-party manufacturing for product candidates, or to do so on commercially reasonable terms, we may not be able to develop and commercialize our product candidates successfully. Our or a third party’s failure to execute on our manufacturing requirements could adversely affect our business in a number of ways, including but not limited to:
| ● | an inability to initiate or continue preclinical studies or clinical trials of product candidates under development; |
| ● | delay in submitting regulatory applications, or receiving regulatory approvals, for product candidates; |
| ● | loss of the cooperation of a collaborator; |
| ● | subjection of our product candidates to additional inspections by regulatory authorities; |
| ● | requirements to cease distribution or to recall batches of our product candidates; and |
| ● | in the event of approval to market and commercialize a product candidate, an inability to meet commercial demands for our products. |
If third parties on which we depend to conduct our preclinical and clinical studies do not perform as contractually required, fail to satisfy regulatory or legal requirements or miss expected deadlines, our development programs could be delayed with materially adverse effects on our business, financial condition, results of operations and prospects.
We rely on CROs and consultants to design, conduct, supervise and monitor preclinical studies for our product candidates and expect to rely on CROs, clinical data management organizations, institutions, and clinical investigators for the conduct of our future clinical trials. We and our CROs are required to comply with various regulations, including Good Laboratory Practices (“GLP”) and Good Clinical Practices (“GCP”) which are enforced by the FDA, and guidelines of the Competent Authorities of the Member States of the European Economic Area (“EEA”) and comparable foreign regulatory authorities to ensure that the health, safety and rights of patients are protected in clinical development, and that study data integrity is assured. Regulatory authorities ensure compliance with these requirements through periodic inspections of study sponsors, principal investigators and study sites. If we or any of our investigators or CROs fail to comply with applicable requirements, the data generated in our studies may be deemed unreliable and the FDA, the EMA or other comparable foreign regulatory authorities may require us to perform additional studies. We cannot be certain that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our studies comply with such requirements.
Our CROs are not our employees, and except for remedies available to us under our agreements with such CROs, we cannot control whether or not they devote sufficient time and resources to our ongoing studies. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines or if the quality or accuracy of the study data they obtain is compromised due to their failure to adhere to our protocols, regulatory requirements or for other reasons, our preclinical or clinical studies may be extended, delayed or terminated.
Because we have relied and continue to rely on third parties, our internal capacity to perform these functions is limited. Outsourcing these functions involves a risk that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. We currently have a relatively small number of employees, which limits the internal resources we have available to identify and monitor our third-party service providers. To the extent we are unable to identify and successfully manage the performance of third-party service providers in the future, our business may be adversely affected. Even though we carefully manage our relationships with our CROs, there can be no assurance that we will not be impacted by this type of challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
23
If we cannot contract with acceptable third parties on commercially reasonable terms, or at all, or if these third parties do not carry out their contractual duties, satisfy legal and regulatory requirements for the conduct of preclinical or clinical studies or meet expected deadlines, our studies could be delayed and otherwise adversely affected. In all events, we are responsible for ensuring that each of our studies is conducted in accordance with the general investigational plan and protocols for the trial.
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. As a biopharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. Restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate include the following:
| ● | the U.S. federal Anti-Kickback Statute prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or paying remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid. A person or entity can be found guilty of violating the statute without actual knowledge of the statute or specific intent to violate it. The term remuneration has been interpreted broadly to include anything of value. Further, courts have found that if “one purpose” of remuneration is to induce referrals, the federal Anti-Kickback statute is violated. Violations are subject to significant civil and criminal fines and penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs. In addition, a claim submitted for payment to any federal healthcare program that includes items or services that were made as a result of a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act, or FCA. The Anti-Kickback Statute has been interpreted to apply to arrangements between biopharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers, among others, on the other; |
| ● | federal civil and criminal false claims laws, including the FCA and civil monetary penalty laws, impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement or record to avoid, decrease or conceal an obligation to pay money to the federal government. A claim that includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim under the FCA. Manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. The FCA also permits a private individual acting as a “whistleblower” to bring qui tam actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery or settlement. When an entity is determined to have violated the FCA, the government may impose civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs; |
24
| ● | the U.S. federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, and also created federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements or using or making any false or fraudulent document in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity can be found guilty of violating HIPAA fraud provisions without actual knowledge of the statute or specific intent to violate it; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information on certain covered healthcare providers, health plans, and healthcare clearinghouses, known as covered entities, as well as their respective “business associates”, being those independent contractors or agents of covered entities that create, receive, maintain, transmit or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, there may be additional federal, state and non-U.S. laws which govern the privacy and security of health and other personal information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; |
| ● | the U.S. Physician Payments Sunshine Act requires applicable manufacturers of covered drugs, devices, biologics and medical supplies to report annually to the Department of Health and Human Services (“HHS”) information related to direct or indirect payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), other licensed healthcare professionals (such as physician assistants, nurse practitioners, and others), and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members; |
| ● | federal government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner to government programs; |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
| ● | analogous state, local and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payors, including private insurers. Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and certain other healthcare providers or marketing expenditures. Additionally, state and foreign laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
The distribution of biopharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of biopharmaceutical products.
Comparable laws and regulations exist in the countries within the EEA and other countries where we may market our products in the future, including without limitation the United Kingdom, Canada and Brazil. Although in the EEA such laws are partially based upon European Union law, they may vary from country to country. Both healthcare and industry specific, as well as general EU and national laws, regulations and industry codes constrain, for example, our interactions with government officials and healthcare practitioners, and the handling of healthcare data. Non-compliance with any of these laws or regulations could lead to criminal or civil liability.
25
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, reputational harm, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to similar penalties. In addition, the approval and commercialization of any product candidate we develop outside the United States will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws. All of these could harm our ability to operate our business and our financial results.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA or EMA regulations or similar regulations of other foreign regulatory authorities, to provide accurate information to the FDA, the EMA or other foreign regulatory authorities, to comply with certain manufacturing standards, to comply with U.S. federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted and implemented a Code of Business Conduct and Ethics, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity, such as employee training on enforcement of the Code of Business Conduct and Ethics, may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions and any imposition of significant fines or other sanctions could have a significant impact on our business and results of operations.
Our collaboration partnerships with pharmaceutical companies are important to our business. If these companies do not successfully develop drugs pursuant to these agreements, our business could be adversely affected.
Under the amended and restated research and collaboration agreement which we entered into with Lilly in December 2022, Lilly has been granted certain exclusive and non-exclusive rights to our Axiomer platform IP to exploit compounds and develop and commercialize products based on this platform. Should Lilly decide not to pursue or be unable to pursue the further development and subsequent commercialization, we would not be able to receive milestone payments or royalty revenues associated with this partnership, which could have a substantial impact on our revenue.
26
We may not succeed in finding and entering into one or more strategic partnerships for our ophthalmology assets QR-504 and QR-1123 on the terms that are attractive to us or at all.
As part of our strategy to focus our efforts on developing our RNA editing platforms, and in addition to having sold the assets sepofarsen and ultevursen in December 2023, we intend to seek one or more third parties to collaborate with respect to the development of our remaining ophthalmology assets, or otherwise sell or transfer such assets in order to generate near-term value. These strategic transactions may be structured in a variety of ways, including receipt of upfront payments, license fees, milestone payments and downstream royalties. However, there can be no assurance that we will be able to identify such a strategic partner or enter into such a transaction with them, or to otherwise sell or transfer our remaining ophthalmology assets. If we fail to do so, we would not receive any such payments related to these assets or otherwise derive any near-term value from them. Even if we are successful in finding a partner for these assets, it is uncertain if such partner will be able to conduct clinical trials meeting its clinical endpoints, obtain regulatory approval and market access in relevant countries. If the partner does not succeed in commercializing the products, we would not receive net sales royalties on such product.
Risks Related to Our Intellectual Property
We may become involved in legal proceedings with third parties that infringe our intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
We may from time to time become involved in legal proceedings challenging our intellectual property rights, including with respect to Axiomer. For example, one or more third parties may seek to invalidate these intellectual property rights. Our success will depend in part on our ability to obtain, maintain and enforce patent protection for our inventions. We may not be able to successfully prosecute the patent applications. If patents are issued or granted, we may fail to maintain these patents due to oppositions and/or other post grant procedures initiated by third parties, we may determine not to pursue litigation against other companies that are infringing these patents, or we may pursue litigation in a variety of ways. For example, we currently are facing oppositions in Japan and Europe with respect to patents relating to our Axiomer platform. If we are unsuccessful in defending our patents against these oppositions, we could lose patent protection for Axiomer and product candidates developed thereunder in these jurisdictions. Further, intellectual property rights may not provide us with complete exclusivity in the respective fields, which would allow for third parties to develop competing products and technologies, which could adversely affect our business and harm our prospects.
We have licensed patent rights from third-party owners or licensees for our ophthalmology programs and if we fail to comply with our obligations in our intellectual property licenses, we could lose rights that are a prerequisite for partnering the ophthalmology assets.
We are party to licenses to third-party intellectual property for certain product candidates that we are seeking third party partners to continue their development. For adRP, we had a world-wide exclusive license to patent rights owned by Ionis Pharmaceuticals, Inc. for the commercial use of gapmers that target mutated Rhodopsin (P23H) mRNA, which has been terminated effective January 2024. For Fuchs Endothelial Corneal Dystrophy, we have a world-wide exclusive sublicensable and royalty-bearing license to patent rights owned by Vico Therapeutics B.V. for the prophylactic and therapeutic use of antisense oligonucleotides for the treatment of FECD.
Our licensing arrangements impose diligence, development, regulatory and commercialization milestones, and royalty, insurance and other obligations on us. If we fail to comply with these obligations, including with respect to programs that are part of the current ophthalmology portfolio that we have suspended as part of the strategy update announced in August 2022, licensors may seek to and have the right to terminate these agreements, which may hamper our ability to find a strategic partner for these programs or may lead to other adverse consequences or payment obligations under the agreements. Such an occurrence could materially adversely affect the value of the product candidate being developed under any such agreement, which would in turn affect our ability to find a strategic partner.
27
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the United States Patent and Trademark Office (“U.S. PTO”) the European Patent Office (“EPO”) and other foreign patent agencies in several stages over the lifetime of the patent. The U.S. PTO, EPO and various foreign national or international patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be rectified by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of patent rights include, but are not limited to, failure to timely file national and regional stage patent applications based on our international patent application, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering our product candidates, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We or our licensors or any future collaborators or strategic partners may become subject to third party claims or litigation alleging infringement of patents or other proprietary rights or seeking to invalidate patents or other proprietary rights, and we may need to resort to litigation to protect or enforce our patents or other proprietary rights, all of which could be costly, time consuming, delay or prevent the development and commercialization of our product candidates, or put our patents and other proprietary rights at risk.
We or our licensors or any future collaborators or strategic partners may be subject to third-party claims for infringement or misappropriation of patent or other proprietary rights. We are generally obligated under our license or collaboration agreements to indemnify and hold harmless our licensors or collaborators for damages arising from intellectual property infringement by us. If we or our licensors, or any future collaborators or strategic partners, are found to infringe a third-party patent or other intellectual property rights, we could be required to pay damages, potentially including treble damages, if we are found to have willfully infringed. In addition, we or our licensors, or any future collaborators or strategic partners, may choose to seek, or be required to seek, a license from a third party, which may not be available on acceptable terms, if at all. Even if a license can be obtained on acceptable terms, the rights may be non-exclusive, which could give our competitors access to the same technology or intellectual property rights licensed to us. If we fail to obtain a required license, we or any future collaborator may be unable to effectively market product candidates based on our technology, which could limit our ability to generate revenue or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations. In addition, we may find it necessary to pursue claims or initiate lawsuits to protect or enforce our patent or other intellectual property rights. The cost to us in defending or initiating any litigation or other proceeding relating to patent or other proprietary rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention for a significant amount of time. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts and limit our ability to continue our operations.
28
Further, if we were to initiate legal proceedings against a third party to enforce a patent covering one of our products or our technology, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the U.S. and in most European countries, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, for example, lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on one or more of our products or certain aspects of our platforms’ technology. Such a loss of patent protection could have a material adverse impact on our business. Patents and other intellectual property rights also will not protect our technology if competitors design around our protected technology without legally infringing our patents or other intellectual property rights.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our business depends upon our ability to develop, manufacture and, if approved, market and sell our product candidates, and to use our proprietary technologies without infringing the proprietary rights of third parties. We cannot guarantee that none of our product candidates, their manufacture, use, sale, offer for sale, or importation into the United States or into any country of the EEA will be held to infringe a third-party patent. As a result, we may become party to, or be threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology, including litigation in a Federal District court, or an interference or a post grant proceeding before the U.S. PTO or litigation in foreign courts or proceedings before foreign patent offices. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future.
Furthermore, in the event a thus far unidentified third party were to assert an infringement claim against us and we were ultimately found to infringe the third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and commercializing our products and technology. However, we may not be able to obtain an appropriate license on commercially reasonable terms or at all. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, in any such proceeding or litigation, we could be found liable for monetary damages. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Any claims by third parties that we have misappropriated their confidential information or trade secrets could have a similar negative impact on our business.
If the third parties from whom we license patent rights do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, or if they retain or license to others any competing rights, our competitive position and business prospects may be adversely affected.
Our success will further depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property, regardless of the exclusive or non-exclusive nature of these rights. Our licensors may not successfully prosecute the patent applications licensed to us. Even if patents are issued or granted, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue litigation less aggressively than we would. Further, the license agreements may not provide us with a complete freedom to operate in the respective fields, which would allow for third parties to develop competing products. Without protection for, or exclusive right to, the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects.
29
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, contract manufacturers, vendors and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, we cannot guarantee that we have executed these agreements with each party that may have or has had access to our trade secrets.
Any party with whom we have executed a confidentiality agreement may breach that agreement and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they disclose such trade secrets, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third-party, our competitive position could be harmed.
We may be subject to claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of our employees’ or consultants’ former employers or their clients. These claims may be costly to defend and if we do not successfully do so, we may be required to pay monetary damages and may lose valuable intellectual property rights or personnel.
Many of our employees were previously employed at universities or biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims in the future that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could compromise our ability to commercialize, or prevent us from commercializing, our product candidates, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may be able to export otherwise infringing products to territories where we have patent protection but where enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. For example, the Special 301 Report (April 2022) from the Office of the United States Trade Representative identified a number of countries, including India and China, where challenges to the procurement and enforcement of patent rights have been reported. Several countries, including India and China, have been listed in the report every year since 1989. As a result, proceedings to enforce our patent rights in certain foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
30
Risks Related to the Commercialization of Our Product Candidates
If any of our product candidates are approved for marketing and commercialization and we are unable to develop sales, marketing and distribution capabilities on our own or enter into agreements with third parties to perform these functions on acceptable terms, we will be unable to successfully commercialize any such future products.
Even if any of our product candidates are approved, we currently have no sales, marketing or distribution capabilities or experience. In addition, we intend to consider out-licensing, spinouts or collaborative partnerships to develop and commercialize our RNA technologies or programs. If any of our product candidates are approved, we will need to enter into collaborations with third parties to provide sales, marketing and distribution capabilities to commercialize such products or develop these capabilities internally, which would be expensive and time-consuming. If we decide to rely on third parties with such capabilities to market our products or decide to co-promote products with collaborators, we will need to establish and maintain marketing and distribution arrangements with such third parties, and there can be no assurance that we will be able to enter into such arrangements on acceptable terms or at all. In entering into third-party marketing or distribution arrangements, any revenue we receive will depend upon the efforts of the third parties and there can be no assurance that such third parties will have adequate sales and distribution capabilities for our products or be successful in gaining market acceptance of any approved product. If we decide to market our products directly, we would need to commit significant financial and managerial resources to develop a marketing and sales force with technical expertise and supporting distribution, administration and compliance capabilities. If we are not successful in commercializing any product approved in the future, either on our own or through third parties, our business, financial condition, results of operations and prospects could be materially adversely affected.
We face competition from entities that have developed or may develop product candidates for our target indications. If these companies develop technologies or product candidates more rapidly than we do or their technologies, including delivery technologies, are more effective, our ability to develop and successfully commercialize. whether on our own or with a collaborator, our product candidates may be adversely affected.
The development and commercialization of drugs is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized biotechnology companies, as well as technology being developed at universities and other research institutions. Our competitors have developed, are developing or may develop product candidates and processes competitive with our product candidates. Competitive therapeutic treatments include those that have already been approved and accepted by the medical community and any new treatments that may enter the market. We believe that a significant number of products are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may try to develop product candidates. An overview of potential competitors is included in Item 4.B: “Business overview - Competition”.
Many of our competitors possess significantly greater financial, technical, manufacturing, marketing, sales and supply resources or experience than us. If we successfully obtain approval for any product candidate, we may face competition based on many different factors, including the safety and effectiveness of our products, the ease with which our products can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these products, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position. Competing products could present superior treatment alternatives, including being more effective, safer, less expensive or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan. Furthermore, in our clinical studies we could be competing for the same patient population, resulting in a smaller population of suitable candidates and possibly delays and additional costs.
31
Even if we are able to commercialize any of our product candidates, whether on our own or with a collaborator, the products may not receive coverage and adequate reimbursement from third-party payors, which could harm our business.
The availability of reimbursement by governmental and private payors is essential for most patients to be able to afford expensive treatments. Sales of any of our product candidates, if approved, by us or a collaborator will depend substantially on the extent to which the costs of these product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors. If reimbursement is not available, or is available only to limited levels, we or our collaborators may not be able to successfully commercialize any product candidate. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services (“CMS”), an agency within the U.S. Department of Health and Human Services, as CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare. Private payors tend to follow CMS to a substantial degree. It is difficult to predict what CMS will decide with respect to reimbursement for fundamentally novel products such as ours, as there is no body of established practices and precedents for these new products. Moreover, reimbursement agencies in Europe may be more conservative than CMS. For example, a number of cancer drugs have been approved for reimbursement in the United States and have not been approved for reimbursement in certain European countries. Factors payors consider in determining reimbursement are based on whether the product is: (i) a covered benefit under its health plan; (ii) safe, effective and medically necessary; (iii) appropriate for the specific patient; (iv) cost-effective; and (v) neither experimental nor investigational.
The intended use of a drug product by a physician can also affect pricing. For example, CMS could initiate a National Coverage Determination administrative procedure, by which the agency determines which uses of a therapeutic product would and would not be reimbursable under Medicare. This determination process can be lengthy, thereby creating a long period during which the future reimbursement for a particular product may be uncertain.
Outside the United States, particularly in Member States of the European Union, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations or the successful completion of health technology assessment procedures with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Certain countries allow companies to fix their own prices for medicines, but monitor and control company profits. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union Member States and parallel distribution, or arbitrage between low-priced and high-priced Member States, can further reduce prices. In some countries, we or our collaborators may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our RNA technology candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of any product candidate approved for marketing is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, financial condition, results of operations or prospects could be adversely affected.
Moreover, efforts by governmental and other third-party payors, in the United States and abroad, to cap or reduce healthcare costs may cause such organizations to limit both coverage and level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale, whether by us or a collaborator, of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
32
Even if we receive marketing approval for any of our product candidates, they may not achieve broad market acceptance, which would limit the revenue that we generate from its sales.
The commercial success of any of our product candidates, if approved by the FDA, the EMA or other regulatory authorities, will depend upon the awareness and acceptance of the product among the medical community, including physicians, patients, patient advocacy groups and healthcare payors. Market acceptance of any of our product candidates, if approved, will depend on a number of factors, including, among others:
| ● | our product candidates’ demonstrated ability to treat patients and to provide patients with incremental therapeutic benefits, as compared with other available treatments; |
| ● | the relative convenience and ease of administration of our product candidates, including as compared with other treatments for patients; |
| ● | the prevalence and severity of any adverse side effects; |
| ● | limitations or warnings contained in the labeling approved for our product candidates by the FDA, the EMA or other regulatory authorities; |
| ● | availability of alternative treatments; |
| ● | pricing and cost effectiveness; |
| ● | the effectiveness of our sales and marketing strategies or those of our collaborators; |
| ● | our, or our collaborators’, ability to increase awareness of our product candidates through marketing efforts; |
| ● | our ability to obtain sufficient third-party coverage or reimbursement; and |
| ● | the willingness of patients to pay out-of-pocket in the absence of third-party coverage. |
Recently enacted and future legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain marketing approval of, and commercialize, our product candidates and may negatively affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could impact our ability to sell our product candidates profitably, if approved.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (the “Medicare Modernization Act”) established the Medicare Part D program and provided authority for limiting the number of drugs that will be covered in any therapeutic class thereunder. The Medicare Modernization Act, including its cost reduction initiatives, could decrease the coverage and reimbursement rate that we or a collaborator receive for any of our approved products. Furthermore, private payors often follow Medicare coverage policies and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from the Medicare Modernization Act may result in a similar reduction in payments from private payors.
33
In March 2010, the Affordable Care Act (“ACA”) was enacted, which substantially changed the way health care is financed by both governmental and private insurers, and significantly impacted the U.S. biopharmaceutical industry. The ACA imposes a significant annual fee on companies that manufacture or import branded prescription drug products. It also contains substantial new provisions intended to broaden access to health insurance, reduce or constrain the growth of health care spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, and impose additional health policy reforms, any of which could negatively impact our business. Among other things, it subjects biological products to potential competition by lower-cost biosimilars, expands the types of entities eligible for the 340B drug discount program, introduced a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of certain branded prescription drugs, and created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. The ACA is likely to continue the downward pressure on pharmaceutical and medical device pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
Since its enactment, there have been numerous judicial, administrative, executive, and legislative challenges to certain aspects of the ACA, including efforts to repeal or replace the ACA. For example, on June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA.
In addition, other legislative and regulatory changes have been proposed and adopted in the United States since the ACA was enacted. For example:
| ● | The U.S. Budget Control Act of 2011, among other things, included aggregate reductions of Medicare payments to providers of 2% per fiscal year. The reductions remain in effect through 2031. |
| ● | The U.S. American Taxpayer Relief Act of 2012 among other things, further reduced Medicare payments to several types of providers. |
| ● | The American Rescue Plan Act of 2021 eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024. |
| ● | Due to the Statutory Pay-As-You-Go Act of 2010, estimated budget deficit increases resulting from the American Rescue Plan Act of 2021, and subsequent legislation, Medicare payments to providers will be further reduced starting in 2025 absent further legislation. |
These laws and regulations may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices we may obtain for any of our product candidates for which we may obtain regulatory approval or the frequency with which any such product candidate is prescribed or used.
Additionally, there has been increasing legislative and enforcement interest in the United States with respect to drug pricing practices. There has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, and review the relationship between pricing and manufacturer patient programs.
34
Further, the government has issued regulations and guidance for states to build and submit importation plans for drugs from outside the United States, pursuant to section 804 of the MMA. CMS has stated that drugs imported by states under this rule will not be eligible for federal rebates under Section 1927 of the Social Security Act and manufacturers would not report these drugs for “best price” or Average Manufacturer Price purposes. Since these drugs are not considered covered outpatient drugs, CMS further stated it will not publish a National Average Drug Acquisition Cost for these drugs. If further implemented, importation of drugs from Canada could materially and adversely affect the price we receive for any of our product candidates.
Additionally, on November 30, 2020, HHS published a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. Pursuant to court order, the removal and addition of the aforementioned safe harbors were delayed, and the inflation Reduction Act of 2022 delayed implementation of the rule until January 1, 2032.
Although a number of these and other proposed measures may require authorization through additional legislation to become effective, and both the Biden administration and Congress have indicated that they will continue to seek new legislative and/or administrative measures to control drug costs.
In addition, the Inflation Reduction Act of 2022 (“IRA”) includes several provisions that will impact our business to varying degrees, including provisions that reduce the out-of-pocket cap for Medicare Part D beneficiaries to $ 2,000 starting in 2025; impose new manufacturer financial liability on certain drugs in Medicare Part D, allow the U.S. government to negotiate Medicare Part B and Part D price caps for certain high-cost drugs and biologics without generic or biosimilar competition, require companies to pay rebates to Medicare for certain drug prices that increase faster than inflation, and delay the rebate rule that would limit the fees that pharmacy benefit managers can charge. The implementation of the IRA is currently subject to ongoing litigation challenging the constitutionality of the Medicare drug price negotiation program. The effects of the IRA on our business and the healthcare industry in general are not yet known.
We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we, or our collaborators, would receive for any approved drug, which could have an adverse effect on customers for our product candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors.
Further, legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals, if any, of our product candidates, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing conditions and other requirements.
Individual states in the U.S. have also become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our products or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
35
In the European Union, similar political, economic and regulatory developments may affect our, or our collaborators’, ability to profitably commercialize our current or any future products. In addition to continuing pressure on prices and cost containment measures, legislative developments at the European Union or Member State level may result in significant additional requirements or obstacles that may increase our operating costs. The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than European Union, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most European Union Member States have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing European Union and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our, or our collaborators’, ability to commercialize any products for which we obtain marketing approval. In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our products. Such reforms could have an adverse effect on anticipated revenue from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop product candidates.
Even if any of our product candidates receive regulatory approval, they may still face future development and regulatory difficulties and we will be subject to extensive post-approval regulatory requirements and review.
If we obtain regulatory approval for any of our product candidates, we and any approved product candidates would be subject to extensive ongoing requirements and review by the FDA, the EMA and foreign regulatory authorities governing the manufacture, quality control, further development, labeling, packaging, storage, distribution, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting of safety and other post-market information. The safety profile of any product will continue to be closely monitored by the FDA, the EMA and comparable foreign regulatory authorities after approval. If the FDA, the EMA or comparable foreign regulatory authorities become aware of new safety information after approval of any of our product candidates, these regulatory authorities may require labeling changes or establishment of a risk management strategy, impose significant restrictions on a product’s indicated uses or marketing, impose ongoing requirements for potentially costly post-approval studies or post-market surveillance or impose a recall.
In addition, manufacturers of therapeutic products and their facilities are subject to continual review and periodic inspections by the FDA, the EMA and other regulatory authorities for compliance with cGMP regulations. For certain commercial prescription drug products, manufacturers and other parties involved in the supply chain must also meet chain of distribution requirements and build electronic, interoperable systems for product tracking and tracing and for notifying the FDA of counterfeit, diverted, stolen and intentionally adulterated products or other products that are otherwise unfit for distribution in the United States. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may, among other things:
| ● | issue untitled letters or warning letters; |
| ● | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
36
| ● | require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| ● | seek an injunction or impose civil or criminal penalties or monetary fines; |
| ● | suspend or withdraw regulatory approval; |
| ● | suspend any ongoing clinical studies; |
| ● | refuse to approve pending applications or supplements to applications filed by us; |
| ● | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
| ● | seize or detain products, refuse to permit the import or export of products, or require us to initiate a product recall. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize our products or generate revenue.
We are subject to anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations are subject to anti-corruption laws, including the U.K. Bribery Act 2010 (“Bribery Act”), the U.S. Foreign Corrupt Practices Act (“FCPA”) and other anti-corruption laws that apply in countries where we do business and may do business in the future. The Bribery Act, the FCPA and other similar laws generally prohibit us, our officers, and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. If we were to pursue sales, marketing or distribution activities in the future, we may operate in a number of jurisdictions that pose a high risk of potential Bribery Act or FCPA violations, and we may participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the Bribery Act, the FCPA or local anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We are also subject to other laws and regulations governing our international operations, including regulations administered by the governments of the United Kingdom and the United States, and authorities in the European Union, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations, collectively referred to as the Trade Control laws, and anti-money laundering laws.
There is no assurance that we will be completely effective in ensuring our compliance with all applicable anti-corruption laws, including the Bribery Act, the FCPA or other legal requirements, including Trade Control laws. If we are not in compliance with the Bribery Act, the FCPA, other anti-corruption laws, Trade Control laws or anti-money laundering laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity. Likewise, any investigation of any potential violations of the Bribery Act, the FCPA, other anti-corruption laws, Trade Control laws or anti-money laundering laws by U.K., U.S. or other authorities could also have an adverse impact on our reputation, our business, results of operations and financial condition.
37
If we receive regulatory approvals and were to pursue sale, marketing or distribution activities ourselves, we may market our product candidates in multiple jurisdictions where we have limited or no operating experience and may be subject to increased business and economic risks that could affect our financial results.
If we receive regulatory approvals, we may market our product candidates ourselves in jurisdictions where we have limited or no experience in marketing, developing and distributing our products. Certain markets have substantial legal and regulatory complexities that we may not have experience navigating. We would be subject to a variety of risks inherent in doing business internationally, including:
| ● | risks related to the legal and regulatory environment in non-U.S. jurisdictions, including with respect to privacy and data security, and unexpected changes in laws, regulatory requirements and enforcement; |
| ● | burdens of complying with a variety of foreign laws in multiple jurisdictions; |
| ● | potential damage to our brand and reputation due to compliance with local laws; |
| ● | fluctuations in currency exchange rates; |
| ● | political, social or economic instability; |
| ● | difficulty of effective enforcement of contractual provisions in local jurisdictions; |
| ● | differences in local business culture and practices, difference in customer attitudes and tendencies and general differences in culture and local trends; |
| ● | the potential need to recruit and work through local partners; |
| ● | reduced protection for or increased violation of intellectual property rights in some countries; |
| ● | inadequate protection against unfair commercial use; |
| ● | difficulties in managing global operations and legal compliance costs associated with multiple international locations; |
| ● | compliance with the Bribery Act, the FCPA, the OECD Convention on Combating Bribery of Foreign Public Officials in International Business Transactions, and similar laws in other jurisdictions; |
| ● | natural disasters, including earthquakes, tsunamis and floods; |
| ● | inadequate local infrastructure; |
| ● | compliance with Trade Control laws and anti-money laundering laws; |
| ● | the effects of applicable foreign tax structures and potentially adverse tax consequences; and |
| ● | exposure to local banking, currency control and other financial-related risks. |
38
The FDA, the EMA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. If we are found to have improperly promoted off-label uses, we may become subject to significant liability.
The FDA, the competent authorities of the EU Member States and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products if approved. In particular, a product may not be promoted for uses that are not approved by the FDA, the EMA or other similar regulatory agencies as reflected in the product’s approved labeling. If we receive marketing approval for our product candidates, physicians may nevertheless prescribe them to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability.
In the United States, engaging in impermissible promotion of our products for off-label uses can also subject us to false claims litigation under federal and state statutes, which can lead to civil and criminal penalties and fines and agreements that materially restrict the manner in which we promote or distribute those products. These false claims statutes include the federal False Claims Act, which allows any individual to bring a lawsuit against a pharmaceutical or medical device company on behalf of the federal government alleging submission of false or fraudulent claims, or causing to present such false or fraudulent claims, for payment by a federal program such as Medicare or Medicaid. If the government prevails in the lawsuit, the individual will share in any fines or settlement funds. Since 2004, these False Claims Act lawsuits against pharmaceutical and medical device companies have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements based on certain sales practices promoting off-label drug uses. This growth in litigation has increased the risk that a company will have to defend a false claim action, pay settlement fines or restitution, agree to comply with burdensome reporting and compliance obligations, and be excluded from the Medicare, Medicaid, and other federal and state healthcare programs. If we do not lawfully promote our approved products, we may become subject to such litigation and, if we are not successful in defending against such actions, those actions may have a material adverse effect on our business, financial condition and results of operations.
Adverse decisions of tax authorities or changes in tax treaties, laws, rules or interpretations could have a material adverse effect on our business, results of operations, financial condition and cash flows.
The tax laws and regulations in the Netherlands, the jurisdiction of our incorporation and our current resident state for tax purposes, may be subject to change and there may be changes in enforcement of tax law. Additionally, European and other tax laws and regulations are complex and subject to varying interpretations. We cannot be sure that our interpretations are accurate or that the responsible tax authority agrees with our views. If our tax positions are challenged by the tax authorities, we could incur additional tax liabilities, which could increase our costs of operations and have a material adverse effect on our business, financial condition, and results of operations.
Further changes in the tax laws of the jurisdictions in which we operate could arise as a result of the base erosion and profit shifting (“BEPS”) project being undertaken by the Organisation for Economic Co-operation and Development (“OECD”). The OECD, which represents a coalition of member countries that encompass certain of the jurisdictions in which we operate or in which we could choose to set up operations in the future, is undertaking studies and publishing action plans that include recommendations aimed at addressing what they believe are issues within tax systems that may lead to tax avoidance by companies. It is possible that the jurisdictions in which we do business could react to the BEPS initiative or their own concerns by enacting tax legislation that could adversely affect us or our shareholders through increasing our tax liabilities.
39
Risks Related to Our Organization, Structure and Operations
Members of our management board and supervisory board and our principal shareholders and their affiliates have significant control over our company, which will limit other stakeholders’ ability to influence corporate matters and could delay or prevent a change in corporate control.
The holdings of the members of our management board and supervisory board and our principal shareholders and their affiliates, represent significant ownership, in the aggregate, of our outstanding ordinary shares (as set out in “Item 7.A. Major Shareholders”). As a result, these shareholders, if they act together, will be able to influence our management and affairs and control the outcome of matters submitted to our shareholders for approval, including the election of members of our management board and supervisory board and any sale, merger, consolidation, or sale of all or substantially all of our assets. These shareholders may have interests, with respect to their ordinary shares, which are different from other investors and the concentration of voting power among these shareholders may have an adverse effect on the price of our ordinary shares. In addition, this concentration of ownership might adversely affect the market price of our ordinary shares by:
| ● | delaying, deferring or preventing a change of control of our Company; |
| ● | impeding a merger, consolidation, takeover or other business combination involving our Company; or |
| ● | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of our Company. |
Please see “Item 7.A. Major Shareholders” for more information regarding the ownership of our outstanding ordinary shares by our management board and supervisory board and our principal shareholders and their affiliates.
Any inability to attract and retain qualified key management and technical personnel would impair our ability to implement our business plan.
Our success largely depends on the continued service of key management and other specialized personnel. The loss of one or more members of our management board or other key employees or advisors could delay our research and development programs and materially harm our business, financial condition, results of operations and prospects. The relationships that our key managers have cultivated within our industry make us particularly dependent upon their continued employment with us. Moreover, we are dependent on the continued service of our technical personnel because of the highly technical nature of our product candidates and technologies and the specialized nature of the regulatory approval process. Because our management board and key employees are not obligated to provide us with continued service, they could terminate their employment or services with us at any time without penalty, subject to providing any required advance notice. Our future success will depend in large part on our continued ability to attract and retain other highly qualified scientific, technical and management personnel, as well as personnel with expertise in clinical testing, manufacturing, governmental regulation and commercialization. It is noteworthy that for 2023 and onwards, given the increased and exclusive focus on the RNA editing technology platform, the fact that we seek to hire and attract personnel in narrower field puts a higher risk factor on our ability to attract personnel. We face competition for personnel from other companies, universities, public and private research institutions, government entities and other organizations.
40
We will need to maintain a good relationship with our employees to maintain our operations. A deterioration in our relationships with our employees could have an adverse impact on our business.
In April and August 2022, we announced an update on our company’s strategy that included a reduction in force totaling approximately 50% of our workforce. Our decision to engage in this reduction resulted from an assessment of our internal resources needs in light of our updated business strategy. This reduction represented a cost-cutting measure across the organization designed to extend our cash runway and to focus our development efforts exclusively on our RNA editing technology platforms. These actions, concluded in March 2023, have caused substantial uncertainty as to job security for the rest of our employees and could damage employee morale across our organization, harm our recruiting and retention efforts and may distract the attention of our company and management from executing on our business strategy. Maintaining good relationships with our employees and operating effectively and efficiently across our organization are crucial to our operations and our success. If we are unable to successfully maintain such relationships or manage the uncertainty as a result of the reduction in the number of our residual workforce, and the complexity of operations, our business may be adversely affected.
We may experience difficulties in managing our growth and expanding our operations.
We have limited experience in drug development. As our product candidates enter and advance through preclinical studies, we will need to expand our development, regulatory and manufacturing capabilities or contract with other organizations to provide these capabilities for us. In the future, we expect to have to manage additional relationships with collaborators or partners, suppliers and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial and management controls, reporting systems and procedures. We may not be able to implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
Data collection and use in the European Union is governed by restrictive regulations governing the use, processing, and cross-border transfer of personal information.
We are subject to a number of laws and regulations relating to privacy and data protection, including, in relation to the European Union, the General Data Protection Regulation (EU) 2016/679 (the “EU GDPR”) and, following the United Kingdom's (“UK”) withdrawal from the EU, the EU GDPR as incorporated into UK laws (“UK GDPR” and together with the EU GDPR, “GDPR”). As the regulatory focus on privacy issues continues to increase and worldwide laws and regulations concerning the handling of personal data expand and become more complex (including in relation to privacy, the use of artificial intelligence, the operation of digital platforms, and provision of digital services), we expect that potential risks related to data collection and use within the business may intensify. In addition, new laws or regulations governing privacy, security, the use of artificial intelligence, the provision of digital services, and data protection may be introduced which could apply in future. The nature and extent of such additions and changes in data protection laws or regulations, and the application to, or impact they may have on, the Company, is uncertain.
The data protection laws applicable to us impact our ability to collect, augment, analyze, use, transfer and share personal data. For example, the GDPR, among other obligations and requirements: (i) imposes requirements relating to having a legal basis or condition for processing personal data and, where applicable, how an organization can collect and rely upon consent obtained from an individual to process their personal data; (ii) imposes obligations in relation to the provision of information to individuals relating to the processing of their personal data; (iii) requires organizations to notify personal data breaches to competent data protection authorities and/or individuals; (iv) imposes additional requirements in relation to the processing of certain categories of data (i.e. “sensitive information” or special category data), which includes health data, data revealing racial or ethnic origin, and genetic data; (v) requiring data protection impact assessments for high risk processing; and (vi) taking certain measures when engaging third-party processors. The GDPR also grants individuals certain rights, subject to certain limitations, including the rights to request and access a copy of the personal data processed by an organization, to have inaccurate personal data rectified or completed if incomplete, to object to or restrict the processing of their personal data, and to request deletion of personal data.
41
Regulators can impose significant monetary fines for violations of laws and regulations relating to privacy and data protection. For example, non-compliance with certain obligations under the GDPR may result in monetary penalties of up to, the greater of, 4 per cent of the Company’s worldwide turnover in the preceding financial year, or €20 million (£17.5 million in the UK), whichever is higher. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. The GDPR, and other changes in laws or regulations associated with the enhanced protection of personal data, mean that the size of potential fines related to data protection and our costs of providing products and services, could result in changes to our business practices, and could prevent us from offering certain services in jurisdictions in which we operate. Although we have implemented contracts, policies and procedures designed to ensure compliance with applicable laws and regulations, there can be no assurance that our employees, contractors, partners, service providers or agents will not violate such laws and regulations or our contracts, policies and procedures. Additionally, public perception and standards related to the privacy of personal data can shift rapidly, in ways that may affect our reputation, the regulators’ approaches to enforcement of existing laws, or influence regulators to enact regulations and laws that may limit our ability to handle personal data or provide certain products and services.
The GDPR, and other applicable data protection laws, also impose restrictions in relation to the international transfer of personal data. For example, in order to transfer data outside of the European Economic Area to a non-adequate country, the GDPR requires us to enter into an appropriate transfer mechanism, and may require us to take additional steps to ensure an essentially equivalent level of data protection. The European Commission has issued standard contractual clauses for data transfers from controllers or processors in the EU (or otherwise subject to the GDPR) to controllers or processors established outside the EU. The new standard contractual clauses require exporters to assess the risk of a data transfer on a case-by-case basis, including an analysis of the laws in the destination country. The UK is not subject to the European Commission’s new standard contractual clauses but has published a UK-specific transfer mechanism, which enables transfers from the UK. The UK-specific mechanism, the “International Data Transfer Agreement”, requires a similar risk assessment of the transfer as the standard contractual clauses. Further, the EU and United States have adopted its adequacy decision for the EU-U.S. Data Privacy Framework ("Framework"), which entered into force on July 11, 2023. This Framework provides that the protection of personal data transferred between the EU and the United States is comparable to that offered in the EU. This provides a further avenue to ensuring transfers to the United States are carried out in line with GDPR. There has been an extension to the Framework to cover UK transfers to the United States. The Framework could be challenged like its predecessor frameworks. Implementing new or revised transfer mechanisms or ensuring an essentially equivalent protection may involve additional expense and potentially increased compliance risk. In the event a legislator, government, regulator or court imposes additional restrictions on international transfers, there may be operational interruption in the performance of services for customers and internal processing of employee information. Such restrictions may also increase our obligations in relation to carrying out international transfers of personal data, and incur additional expense and increased regulatory liabilities.
Our failure to comply with U.S. data protection laws and regulations could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results.
There are numerous laws and legislative and regulatory initiatives at the U.S. federal and state levels as well addressing privacy and security concerns, and some state privacy laws apply more broadly than HIPAA and associated regulations. For example, the California Consumer Privacy Act (“CCPA”), creates individual privacy rights for California consumers (as defined in the law) and places increased privacy and security obligations on entities handling personal information about residents of California. The CCPA requires covered companies to provide certain disclosures to consumers about its data collection, use and sharing practices, and to provide affected California residents with ways to opt-out of certain sales or transfers of personal information. The CCPA went into effect on January 1, 2020, and became enforceable by the California Attorney General on July 1, 2020. The CCPA provides for civil penalties for violations, as well as a private right of action for certain data breaches that result in the loss of personal information. This private right of action may increase the likelihood of, and risks associated with, data breach litigation. While there is currently an exception for protected health information that is subject to the Health Insurance Portability and Accountability Act of 1996, or HIPAA, and clinical trial regulations, as currently written, the CCPA may impact our business activities. There continues to be uncertainty surrounding the enforcement and implementation of the CCPA, exemplifying the vulnerability of our business to the evolving regulatory environment related to personal data and protected health information.
42
Further, as of January 1, 2023, the California Privacy Rights Act (“CPRA”), amended the CCPA and created additional obligations with respect to processing and storing personal information and sensitive personal information. The effects of the CCPA (as modified by the CPRA) are potentially significant and may require us to modify our data collection or processing practices and policies and to incur substantial costs and expenses in an effort to comply and increase our potential exposure to regulatory enforcement and/or litigation.
Similar consumer privacy laws have passed or come into force in more than a dozen U.S. states. Like the CCPA, these laws grant consumers rights in relation to their personal information and impose new obligations on regulated businesses, including, in some instances, broader data security requirements. In addition, federal and state legislators and regulators have signaled their intention to further regulate health and other sensitive information, and new and strengthened requirements relating to this information could impact our business. At the state level, some states have passed or proposed laws to specifically regulate health information. For example, Washington’s My Health My Data Act, which comes into force in March 2024, requires regulated entities to obtain consent to collect health information, grants consumers certain rights, including to request deletion, and provides for robust enforcement mechanisms, including enforcement by the Washington state attorney-general and a private right of action for consumer claims. At the federal level, the FTC has used its authority over “unfair or deceptive acts or practices” to impose stringent requirements on the collection and disclosure of sensitive categories of personal information, including health information. Moreover, the FTC’s expanded interpretation of a “breach” under its Health Breach Notification Rule could impose new disclosure obligations that would apply in the event of a qualifying breach.
We are increasingly dependent on information technology systems, and our systems and infrastructure face certain risks, including from cyber security breaches and data leakage, which in turn could lead to a determination that we have failed to comply with GDPR, resulting in a fine or regulatory enforcement action.
We increasingly rely upon technology systems and infrastructure, including support provided by our partners and third parties, to support our business. For example, we routinely rely on our technology systems and infrastructure to aid us in the collection, use, storage and transfer, disclosure and other processing of voluminous amounts of data (including confidential, business, personal and other sensitive information). We also rely on systems for manufacturing, regulatory compliance and various other matters.
The increasing use and evolution of technology, including cloud-based computing, and reliance on third parties creates additional opportunities for the unintentional, intentional and/or unauthorized exposure, dissemination, misuse, and/or destruction of confidential information stored in our technology systems, infrastructure, and products. Our computer systems, servers, and other technology systems (and those of third parties that we use) are vulnerable to breakdown, interruption, cyber and other security attacks, system malfunction, unauthorized access, misuse, and other events. Security threats, including cyber and other attacks are becoming increasingly sophisticated, frequent, and adaptive. For example, in February 2021 we have experienced an incident in which personnel information was inadvertently made available to company employees broadly. In response, we made the required reports to regulatory authorities and took remedial measures to secure such information systems from unauthorized and unintentional access. Any such vulnerability could compromise our technology systems and infrastructure and could expose personal and/or proprietary information (including sensitive personal information) to unauthorized third parties and/or cause permanent loss of such data. While we have invested in the protection of data and information technology, there can be no assurance that our efforts will prevent or adequately address breakdowns, breaches in our systems compromises or other incidents or ensure compliance with all applicable security and privacy laws, regulations and standards, including with respect to third-party service providers that utilize sensitive personal information, including protected health information on our behalf. We could also suffer strained relationships, increased costs (for security measures, remediation or otherwise), litigation (including class actions and stockholder derivative actions) or other negative consequences (including a decline in stock price) from breaches, cyber and other security attacks, industrial espionage, ransomware, email or phishing scams, malware or other cyber incidents, which may compromise our system infrastructure or lead to data leakage, either internally or at our third-party providers or other business partners. Some cyber security incidents may be very difficult to prevent or adequately address.
43
Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs, a determination that we have failed to comply with GDPR, resulting in a fine, and adversely affect our business.
Despite the implementation of security measures, our information technology and other internal infrastructure systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, cyberattacks and unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. The risk of cyber incidents could also be increased by cyberwarfare in connection with the ongoing conflict between Russia and Ukraine and the conflict in Israel, including potential proliferation of malware into systems unrelated to such conflicts. A significant disruption in the availability of our information technology and other internal infrastructure systems could cause interruptions in our collaborations with our partners and delays in our research and development work. For instance, the loss of preclinical or clinical data involving our product candidates could result in delays in our development and regulatory filing efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on third parties to manufacture our product candidates and will rely on third parties to conduct future clinical trials, and similar events relating to their computer systems could also have similar consequences to our business. To the extent that any disruption or security breach were to result in unauthorized disclosure of, or access to, or accidental or unlawful loss, destruction, alteration of, or damage to, our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability, including fines for failure to comply with GDPR (to the extent the data contained personal data), and the development of our product candidates could be delayed.
The use of new and evolving technologies, such as artificial intelligence (“AI”), in our offerings may present risks and challenges that can impact our business including by posing security risks to our confidential information, proprietary information, and personal data.
We continue to build and integrate AI into our offerings, and this innovation presents risks and challenges that could affect its adoption, and therefore our business. The use of certain artificial intelligence technology can give rise to intellectual property risks, including compromises to proprietary intellectual property and intellectual property infringement. Additionally, we expect to see increasing government and supranational regulation related to artificial intelligence use and ethics, which may also significantly increase the burden and cost of research, development and compliance in this area. For example, the EU’s Artificial Intelligence Act (“AI Act”) — the world’s first comprehensive AI law — is anticipated to enter into force in 2024 and, with some exceptions, become effective 24 months thereafter. This legislation imposes significant obligations on providers and deployers of high-risk artificial intelligence systems, and encourages providers and deployers of artificial intelligence systems to account for EU ethical principles in their development and use of these systems. If we develop or deploy AI systems that are governed by the AI Act, we may be required to adopt higher standards of data quality, transparency, and human oversight, and adhere to specific and potentially burdensome and costly ethical, accountability, and administrative requirements. The rapid evolution of AI will require the application of significant resources to design, develop, test and maintain our products and services to help ensure that AI is implemented in accordance with applicable law and regulation and in a socially responsible manner and to minimize any real or perceived unintended harmful impacts. Our vendors may in turn incorporate AI tools into their own offerings, and the providers of these AI tools may not meet existing or rapidly evolving regulatory or industry standards, including with respect to privacy and data security. Further, bad actors around the world use increasingly sophisticated methods, including the use of artificial intelligence, to engage in illegal activities involving the theft and misuse of personal information, confidential information, and intellectual property. Any of these outcomes could damage our reputation, result in the loss of valuable property and information, and adversely impact our business.
44
Our current operations are mainly concentrated in one location, and any events affecting this location may have material adverse consequences.
Our current operations are primarily located in our facilities in Leiden, the Netherlands. Any unplanned event, such as flood, fire, explosion, earthquake, extreme weather condition, medical epidemics, power shortage, telecommunication failure or other natural or manmade accidents or incidents that result in us being unable to fully utilize the facilities may have a material adverse effect on our ability to operate our business, particularly on a daily basis, and have significant negative consequences on our financial and operating conditions. Loss of access to these facilities may result in increased costs, delays in the development of our product candidates or interruption of our business operations. As part of our risk management policy, we maintain insurance coverage at levels that we believe are appropriate for our business. However, in the event of an accident or incident at these facilities, we cannot assure you that the amounts of insurance will be sufficient to satisfy any damages and losses. If our facilities are unable to operate because of an accident or incident or for any other reason, even for a short period of time, any or all of our research and development programs may be harmed.
Our international operations subject us to various operational risks, and our failure to manage these risks could adversely affect our results of operations.
We face significant operational risks as a result of doing business internationally, such as:
| ● | fluctuations in foreign currency exchange rates; potentially adverse and/or unexpected tax consequences, including penalties due to the failure of tax planning or due to the challenge by tax authorities on the basis of transfer pricing and liabilities imposed from inconsistent enforcement; |
| ● | potential changes to relevant accounting standards, which may impact our reported financial situation and results; |
| ● | being subject to the different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations; |
| ● | reduced protection of, or significant difficulties in enforcing, intellectual property rights in certain countries; |
| ● | difficulties in attracting and retaining qualified personnel due to the location of our operations; |
| ● | restrictions imposed by local labor practices and laws of jurisdictions where we have employees on our business and operations, including unilateral cancellation or modification of contracts; |
| ● | rapid changes in global government, adverse market or macroeconomic conditions or market volatility resulting from global economic developments, political or civil unrest or instability, including due to war, international hostilities or terrorism (such as the ongoing conflict between Russia and Ukraine and the conflict in Israel), high inflation, the post-COVID environment or future health epidemics and other similar outbreaks or events, and potential failure in confidence of our suppliers or customers due to such changes or events; and |
| ● | tariffs, trade protection measures, import or export licensing requirements, trade embargoes and other trade barriers. |
If we were to experience any of the difficulties listed above, or any other difficulties, our international development activities and our overall financial condition may suffer and cause it to reduce or discontinue our international operations and market approval efforts.
45
Inadequate funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, which in turn could prevent new products from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA and other government employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, future shutdowns of other government agencies, such as the SEC, may also impact our business by delaying review of our public filings, which in turn could delay or frustrate our ability to access the public capital markets. Similar developments at regulators (including the EMA) in other countries could have similar impacts on our applications for marketing approval and on our business.
The investment of our cash and cash equivalents is subject to risks which may cause losses and affect the liquidity of these investments.
As at December 31, 2023, we had € 118,925,000 in cash and cash equivalents. To date, our cash and cash equivalents have been deposited primarily in savings and deposit accounts with original maturities of twelve months or less. Savings and deposit accounts generate small amounts of interest income, if any. In 2021 and 2022, some of our euro savings accounts required us to incur small amounts of interest expenses. In 2023, we have generated interest income on our euro deposit accounts. Any future investments may include term deposits, corporate bonds, money market funds and government securities, all in accordance with our cash management policy. These investments are subject to general credit, liquidity, market and interest rate risks. For example, interest rates may be negative in the European Union. We may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our financial statements.
In addition, should our investments cease paying or reduce the amount of interest paid to us, our interest income would suffer. The materialization of any of the market risks associated with our investment portfolio may have an adverse effect on our results of operations, liquidity and financial condition.
We may be exposed to significant foreign exchange risk.
We incur portions of our expenses, and may in the future derive revenues, in currencies other than our functional currency (i.e. the euro), in particular, the U.S. dollar. As a result, we are exposed to foreign currency exchange risk as our results of operations and cash flows are subject to fluctuations in foreign currency exchange rates. We currently do not engage in hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the euro. Therefore, for example, an increase in the value of the U.S. dollar against the euro could be expected to have a negative impact on our expenditures, although it is our policy to match the currency of our cash and cash equivalents with expected cash out flows as much as practically feasible. We cannot predict the impact of foreign currency fluctuations, and foreign currency fluctuations in the future may adversely affect our financial condition, results of operations and cash flows.
46
Our business entails a significant risk of product liability and our ability to obtain sufficient insurance coverage could have a material effect on our business, financial condition, results of operations or prospects.
The use of our product candidates in preclinical studies, in clinical trials and the sale of any of our product candidates if approved, exposes us to the risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers or others selling or otherwise coming into contact with our product candidates. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, including as a result of interactions with alcohol or other drugs, negligence, strict liability, and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we become subject to product liability claims and cannot successfully defend ourselves against them, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in, among other things:
| ● | withdrawal of patients from our clinical trials; |
| ● | substantial monetary awards to patients or other claimants; |
| ● | decreased demand for any of our current or future product candidates following marketing approval, if obtained; |
| ● | damage to our reputation and exposure to adverse publicity; |
| ● | increased FDA or EMA warnings on product labels; |
| ● | litigation costs; |
| ● | distraction of management’s attention from our primary business; |
| ● | loss of revenue; and |
| ● | the inability to successfully commercialize any of our current or future product candidates, if approved. |
We will need to maintain product liability insurance coverage for our clinical trials. We may not be able to obtain such coverage at a reasonable cost or in sufficient amounts to protect us against losses, including if insurance coverage becomes increasingly expensive. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial, particularly in light of the size of our business and financial resources. A product liability claim or series of claims brought against us could cause our share price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, business and prospects could be materially adversely affected.
Our ability to use our net operating losses to offset future taxable income may be subject to certain limitations.
Our ability to use our net operating losses (“NOLs” or “tax losses”) in the Netherlands is currently limited and may be further limited. Under Dutch income tax law, effective from January 1, 2022, tax losses may be carried forward indefinitely. However, the offset of losses will be limited in a given year against the first € 1.0 million of taxable profit. For taxable profit in excess of this amount, losses may only be offset up to 50% of this excess. As at December 31, 2023, we had a total of € 402.3 million tax loss carry-forwards available for offset against future taxable profits.
There is also a risk that due to regulatory changes such as suspensions on the use for NOLs, our existing NOLs could expire or otherwise become unavailable to offset future income tax liabilities. For these reasons, we may not be able to use a material portion of the NOLs, even if we attain profitability.
47
Changes in accounting rules and regulations, or interpretations thereof, could result in unfavorable accounting charges or require us to change our compensation policies.
Accounting methods and policies for biopharmaceutical companies, including policies governing revenue recognition, research and development and related expenses and accounting for share-based compensation, are subject to review, interpretation and guidance from relevant accounting authorities, as well as the SEC. Changes to accounting methods or policies, or interpretations thereof, may require us to reclassify, restate or otherwise change or revise our financial statements, including those contained in this Annual Report.
Risks Related to Ownership of our Ordinary Shares
We cannot predict what the market price of our ordinary shares will be. As a result, it may be difficult for investors to sell our ordinary shares at or above the price at which they purchased them.
An active trading market for our shares may not be sustained. The market value of our ordinary shares may decrease from time to time. As a result of these and other factors, investors may be unable to resell our shares at or above the price at which they purchased them. The lack of an active market may impair investors’ ability to sell our shares at the time they wish to sell them or at a price that they consider reasonable. The lack of an active market may also reduce the fair market value of our shares. Further, an inactive market may also impair our ability to raise capital by selling our ordinary shares and may impair our ability to enter into strategic partnerships or acquire companies or products by using our ordinary shares as consideration. The market price of our shares may be volatile and investors could lose all or part of their investment.
The trading price of our ordinary shares is likely to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. For example, the price of our ordinary shares, which reached its high record of $ 27.60 per share at the close of the trading on March 16, 2015, decreased as low as $ 0.56 per share at the close of the trading on May 11, 2022. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this Annual Report, these factors include:
| ● | the results of our preclinical studies and clinical trials; |
| ● | the presentation of data at industry conferences by us and/or our competitors; |
| ● | the responses to any of our INDs with the FDA and any of our clinical trial applications with the competent authorities of the EU Member States; |
| ● | any current or future preclinical studies or clinical trials of our product candidates, including any delays in enrollment rates or timing of these trials; |
| ● | regulatory actions with respect to our products or our competitors’ products; |
| ● | the recruitment or departure of key personnel; |
| ● | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments; |
| ● | expiration or termination of licenses, research contracts or other collaboration agreements; |
| ● | the timing and cost of, and level of investment in, research and development activities relating to our product candidates, which may change from time to time; |
| ● | results of clinical trials of our competitors; |
| ● | the success of competitive products or technologies; |
48
| ● | actual or anticipated changes in our growth rate relative to our competitors; |
| ● | regulatory or legal developments in the United States, the European Union and other jurisdictions; |
| ● | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| ● | the level of expenses related to any of our product candidates or preclinical or clinical development programs; |
| ● | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
| ● | variations in our financial results or those of companies that are perceived to be similar to us; |
| ● | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
| ● | share price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
| ● | announcement or expectation of additional financing efforts; |
| ● | sales of our ordinary shares by us, our insiders or our other shareholders; |
| ● | changes in the structure of healthcare payment systems; |
| ● | market conditions in the pharmaceutical and biotechnology sectors; and |
| ● | general economic, industry and market conditions, including the impact of inflation and rising interest rates, and domestic or international political instability. |
In addition, the stock market in general, and pharmaceutical and biotechnology companies in particular, have experienced extreme price and volume fluctuations that have sometimes been unrelated or disproportionate to the operating performance of these companies. Historically, securities class action litigation has often been brought against companies following periods of volatility in the market price of a company’s securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management’s attention and resources, which could harm our business, operating results, or financial condition. Broad market and industry factors may negatively affect the market price of our ordinary shares, regardless of our actual operating performance. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our ordinary shares.
If we are unable to maintain compliance with Nasdaq listing standards, our ordinary shares may be delisted from the Nasdaq Stock Market and you may face significant restrictions on the resale of your shares.
There can be no assurances that we will be able to maintain our Nasdaq listing in the future. For example, in May 2022, we received a notification from Nasdaq that the trading price of our ordinary shares did not meet the minimum bid price requirement of $ 1.00 per share for a period of 30 consecutive trading days. We were granted a 180-day compliance period to regain compliance by achieving a closing bid price of at least $ 1.00 per share for a minimum of 10 consecutive trading days. In October 2022, we transferred our listing from the Nasdaq Global Market to the Nasdaq Capital Market and received an additional 180-day compliance period, as contemplated by Nasdaq. On December 2, 2022, Nasdaq informed us that we had regained compliance with the minimum bid price rule. Nevertheless, share prices in biotech are volatile and there can be no assurances that we will be able to maintain our Nasdaq Capital Market listing in the future.
49
In the event we are unable to maintain compliance with the Nasdaq Capital Market listing standards and our ordinary shares are delisted from the exchange, it would, among other things, likely lead to a number of negative implications, including an adverse effect on the price of our ordinary shares, reduced liquidity in our ordinary shares, the loss of federal preemption of state securities laws, breach of covenants in agreements and greater difficulty in obtaining financing. In the event of a de-listing, we would take actions to restore our compliance with Nasdaq’s listing requirements, but we can provide no assurance that any such action taken by us would allow our ordinary shares to become listed again, stabilize the market price or improve the liquidity of our ordinary shares, prevent our ordinary shares from dropping below the Nasdaq minimum bid price requirement in the future, or prevent future non-compliance with Nasdaq’s listing requirements. If we cannot restore our compliance with Nasdaq’s listing requirements, we would pursue eligibility for trading of these securities on other markets or exchanges, such as the OTCQB or OTCQX, which are unorganized, inter-dealer, over-the-counter markets which provides significantly less liquidity than the Nasdaq Capital Market or other national securities exchanges.
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and trading price of our ordinary shares.
The global credit and financial markets have experienced extreme volatility and disruptions in the past several years, most recently due to the COVID-19 pandemic, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates and uncertainty about economic stability. We believe that the state of global economic conditions remain particularly volatile and uncertain, the impact of fiscal stimulus programs initiated by various governments in response to the pandemic and economic downturn and ongoing supply chain issues and inflationary pressures, but also due to recent and expected shifts in political, legislative and regulatory conditions concerning, among other matters, international trade and taxation, and that an uneven recovery or a renewed global downturn may negatively impact our ability to conduct clinical trials on the scale and timelines anticipated. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions, whether due to the effects of the COVID-19 pandemic or otherwise, will not occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business or political environment or continued unpredictable and unstable market conditions. The financial markets and the global economy may also be adversely affected by the current or anticipated impact of military conflict, including the conflict between Russia and Ukraine and the conflict in Israel, terrorism or other geopolitical events. Sanctions imposed by the United States and other countries in response to such conflicts, including the conflict in Ukraine, may also adversely impact the financial markets and the global economy, and any economic countermeasures by the affected countries or others could exacerbate market and economic instability. If the current equity and credit markets deteriorate, it may make obtaining any necessary debt or equity financing more difficult, more costly and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers and other partners may not survive an economic downturn, which could directly affect our ability to attain our operating goals on schedule and on budget. To the extent that our profitability and strategies are negatively affected by downturns or volatility in general economic conditions, our business and results of operations may be materially adversely affected.
If securities or industry analysts publish inaccurate or unfavorable research or cease to publish research about our business, our share price and trading volume could decline.
The trading market for our ordinary shares depends in part on the research and reports that securities or industry analysts publish about us or our business. In the event securities or industry analysts who cover us downgrade our ordinary shares, or publish inaccurate or unfavorable research about our business, our share price would likely decline. In addition, if one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our ordinary shares could decrease, which might cause our share price and trading volume to decline.
50
Sales of a substantial number of our ordinary shares by our existing shareholders in the public market could cause our share price to fall.
If our existing shareholders sell, or indicate an intention to sell, substantial amounts of our ordinary shares in the public market, the trading price of our ordinary shares could decline. In addition, a substantial number of ordinary shares subject to outstanding options are or will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules. If these additional ordinary shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of our ordinary shares could decline.
If we sell our ordinary shares in future financings, shareholders may experience immediate dilution and, as a result, our share price may decline.
We may from time to time issue additional ordinary shares or securities convertible into our ordinary shares, including in future financings that we may undertake. For example, in December 2022, we issued 9,381,586 shares to Lilly pursuant to our expanded licensing and research collaboration with Lilly, resulting in gross proceeds of € 14,122,000. In September 2021, we issued 3,989,976 shares to Lilly pursuant to the original collaboration, resulting in gross proceeds of € 25,270,000. In April 2021, we consummated an underwritten public offering of 15,923,077 ordinary shares at an issue price of $ 6.50 per share. The gross proceeds from this offering amounted to € 88,115,000.
In November 2021, we entered into a sales agreement for at-the-market offerings up to a maximum aggregate offering price of $ 75,000,000 of our ordinary shares with Cantor Fitzgerald & Co.
In September 2022, we extinguished our debt with Pontifax Medison Finance and Kreos Capital by repaying all outstanding principal amounts and interest accrued thereon. In connection with our prior loan facility with Pontifax and Kreos, we issued warrants to purchase up to an aggregate of 679,628 ordinary shares, with exercise prices ranging between $ 7.00 and $ 12.00. These warrants remain in place until their five-year economic life expires.
Due to the issuances described above and any future additional issuances of shares of our common stock or securities convertible into common stock, including pursuant to our shelf registration statement or our ATM facility, our shareholders may experience immediate dilution and, as a result, our stock price may decline.
Because we do not anticipate paying any cash dividends on our ordinary shares in the foreseeable future, capital appreciation, if any, will be the sole source of potential gain for our shareholders.
We have never declared or paid cash dividends on our ordinary shares. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. As a result, capital appreciation, if any, of our ordinary shares will be the sole source of gain for our shareholders for the foreseeable future.
We have been a listed company since September 2014. Complying with all requirements will increase our costs, require additional management resources and qualified accounting and financial personnel, and we may fail to meet all of these obligations.
We face substantial legal, accounting, administrative and other costs and expenses as a public company. Compliance with the Sarbanes-Oxley Act of 2002, the Dodd-Frank Act of 2010, the Dutch Financial Supervision Act and the rules promulgated thereunder, as well as rules of the SEC and Nasdaq and the Dutch Corporate Governance Code (“DCGC”) for example, are expected to result in ongoing increases in our legal, audit and financial compliance costs. The Securities Exchange Act of 1934, as amended (the “Exchange Act”) requires, among other things, that we file certain periodic reports with respect to our business and financial condition. We must also comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002. Complying with Section 404 is costly and management’s attention may be diverted from other business concerns, which could adversely affect our business and results of operations.
51
We currently do not have an internal audit group, and we will need to continue to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge, and it may be difficult to recruit and maintain such personnel. Implementing any appropriate changes to our internal controls may require specific compliance training for our directors, officers and employees, entail substantial costs to modify our existing accounting systems, and take a significant period of time to complete. Such changes may not, however, be effective in maintaining the adequacy of our internal controls, and any failure to maintain that adequacy, or consequent inability to produce accurate financial statements or other reports on a timely basis, could increase our operating costs and could materially impair our ability to operate our business.
If we fail to establish and maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, shareholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our ordinary shares.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act of 2002, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our ordinary shares.
Our management board is required to assess the effectiveness of our internal controls and procedures annually and, in case we become a domestic filer, we will be required to disclose changes to these controls on a quarterly basis. Our independent registered public accounting firm is required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to certain reporting requirements of the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosures due to error or fraud may occur and not be detected.
52
Risks Related to Investing in a Foreign Private Issuer or a Dutch Company
We are a foreign private issuer and, as a result, we are not subject to U.S. proxy rules and are subject to Exchange Act reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
We report under the Exchange Act as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act and although we are subject to Dutch laws and regulations with regard to such matters and intend to furnish quarterly financial information to the SEC, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including:
| ● | the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; |
| ● | the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and |
| ● | the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events. |
In addition, foreign private issuers are not required to file their annual report on Form 20-F until 120 days after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, our shareholders may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
Certain U.S. holders of our ordinary shares may suffer adverse tax consequences if we are characterized as a passive foreign investment company for U.S. federal income tax purposes.
Generally, if, for any taxable year, at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company (“PFIC”) for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest, and gains from the sale or exchange of investment property and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. If we are characterized as a PFIC, our U.S. shareholders may suffer adverse tax consequences, including having gains realized on the sale of our ordinary shares treated as ordinary income, rather than capital gain, the loss of the preferential rate applicable to dividends received on our ordinary shares by individuals who are U.S. holders, and having interest charges apply to distributions by us and the proceeds of share sales. See Item 10.E: “Taxation” for more information.
The market value of our assets may be determined in large part by reference to the market price of our ordinary shares, which is likely to fluctuate and may fluctuate considerably given that market prices of technology companies have been especially volatile. In addition, the composition of our income and assets will be affected by how, and how quickly, we spend our cash, including any cash raised pursuant to prior offerings. Based on the average value of our gross assets and composition of our income, we believe that we were a PFIC for the 2023 taxable year. However, our status as a PFIC is a fact-intensive determination made on an annual basis, and we cannot provide any assurance regarding our PFIC status for the current, prior or future taxable years.
We intend to provide the information necessary for U.S. holders to make a “qualified electing fund,” (“QEF”) election if we are treated as a PFIC for any taxable year.
53
The rights and responsibilities of our shareholders are governed by Dutch law and differ in some important respects from the rights and responsibilities of shareholders under U.S. law.
Our corporate affairs are governed by our articles of association, our internal rules and by the laws governing companies incorporated in the Netherlands. The rights of our shareholders and the responsibilities of members of our supervisory board under Dutch law are different than under the laws of some U.S. jurisdictions. In the performance of their duties, our management board and our supervisory board are required by Dutch law to consider the interests of ProQR Therapeutics, its shareholders, its employees and other stakeholders and not only those of our shareholders. Also, as a Dutch company, we are not required to solicit proxies or prepare proxy statements for general meetings of shareholders. Dutch law does not have a regulatory regime for U.S.-style proxy solicitations and, even though Dutch law accommodates voting by proxy, the solicitation of proxies is not a widely used business practice in the Netherlands. In addition, the rights of holders of shares and many of the rights of shareholders as they relate to, for example, the exercise of shareholder rights, are governed by Dutch law and our articles of association and differ from the rights of shareholders under U.S. law. For example, Dutch law does not grant appraisal rights to a company’s shareholders who wish to challenge the consideration to be paid upon a merger or consolidation of the company.
We may lose our foreign private issuer status which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
We are a foreign private issuer and therefore we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. In order to maintain our current status as a foreign private issuer, either:
| ● | a majority of our ordinary shares must be either directly or indirectly owned of record by non-residents of the United States; or |
| ● | a majority of our “executive officers” and a majority of our directors may not be U.S. citizens or residents, more than 50% of our assets cannot be located in the United States and our business must be administered principally outside the United States. |
If we lose this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers.
We may also be required to make changes in our corporate governance practices in accordance with various SEC and Nasdaq rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the costs we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs, would make some activities more time consuming and costly and could necessitate the hiring of additional accounting, financial and legal staff. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our supervisory board.
We currently report our financial results under IFRS, which differ in certain significant respects from U.S. GAAP.
Currently we report our financial statements under International Financial Reporting Standards (“IFRS”). There have been and there may in the future be certain significant differences between IFRS and U.S. GAAP, including differences related to revenue recognition, share-based compensation expense, income tax and earnings per share. As a result, our financial information and reported earnings for historical or future periods could be significantly different if they were prepared in accordance with U.S. GAAP. In addition, we do not intend to provide a reconciliation between IFRS and U.S. GAAP unless it is required under applicable law. As a result, you may not be able to meaningfully compare our financial statements under IFRS with those companies that prepare financial statements under U.S. GAAP.
54
Provisions of our articles of association or Dutch corporate law might deter acquisition bids for us that might be considered favorable and prevent or frustrate any attempt to replace or remove the management board and supervisory board.
Certain provisions of our articles of association may make it more difficult for a third party to acquire control of us or effect a change in our management board or supervisory board. These provisions include:
| ● | the authorization of a class of preferred shares that may be issued against payment of at least 25% of the nominal value thereof to a protection foundation, for which we have granted a perpetual and repeatedly exercisable call option to such protection foundation, for up to such number of preferred shares as equals, at the time of exercise of the call option, the lesser of: (i) the total number of shares equal to our issued share capital at that time minus the number of preferred shares already held by the protection foundation at that time (if any) or (ii) the maximum number of preferred shares that may be issued under our authorized share capital under our articles of association from time to time; |
| ● | a provision that our management board members and supervisory board members may only be appointed upon a binding nomination by our supervisory board, which can be set aside by a two-thirds majority of our shareholders representing more than half of our issued share capital; |
| ● | a provision that our management board members and supervisory board members may only be removed or suspended by our general meeting of shareholders by a two-thirds majority of votes cast representing more than 50% of our issued share capital (unless the removal was proposed by the supervisory board); and |
| ● | a requirement that certain matters, including an amendment of our articles of association, may only be brought to our shareholders for a vote upon a proposal by our management board that has been approved by our supervisory board. |
In addition, under Dutch law, our management board and supervisory board need to act in the interest of the company and our business. The boards are required to take into account the interests of all our stakeholders, including by promoting the sustainable success of our business and the creation of long term value for us and our business. The boards are responsible for determining our strategy and choosing our strategic direction. In doing so and depending on the circumstances they may decide to not entertain a proposed takeover or other strategic proposal, even if the proposal is supported by the majority of our shareholders and/or would create more shareholder value. The boards may also use their general authority under Dutch corporate law and the DCGC to use (ad-hoc) defensive mechanisms against and/or not co-operate with a proposal, e.g., by not providing due diligence and or by not cooperating with shareholder proposals to adopt resolutions in a general shareholder meeting that may change our strategy. The boards may for instance invoke the maximum 180 days response time set out in the DCGC and/or the Dutch statutory reflection period of up to 250 days in response to shareholder activists seeking changes in the composition of the boards and/or upon hostile takeover attempts.
As indicated above, we have adopted an anti-takeover measure by granting a perpetual and repeatedly exercisable call option to the protection foundation, which confers upon the protection foundation the right to acquire, under certain conditions, the number of preferred shares described above. The issuance of such preferred shares will occur upon the protection foundation’s exercise of the call option and will not require shareholder consent. Such a measure has the effect of making a takeover of us more difficult or less attractive and as a result, our shareholders may be unable to benefit from a change of control and realize any potential change of control premium which may materially and adversely affect the market price of our ordinary shares.
55
We are not obligated to and do not comply with all the best practice provisions of the Dutch Corporate Governance Code. This may affect our shareholders’ rights.
As a Dutch company we are subject to the DCGC. The DCGC contains both principles and best practice provisions that regulate relations between the management board, the supervisory board and the shareholders (i.e., the general meeting of shareholders). The DCGC is based on a “comply or explain” principle. Accordingly, companies are required to disclose in their annual reports, filed in the Netherlands, whether they comply with the provisions of the DCGC. If they do not comply with those provisions (e.g., because of a conflicting Nasdaq requirement), we are required to give the reasons for such non-compliance.
The DCGC applies to all Dutch companies listed on a government-recognized stock exchange, whether in the Netherlands or elsewhere, including Nasdaq. We do not comply with all the best practice provisions of the DCGC. This may affect our shareholders’ rights and they may not have the same level of protection as a shareholder in a Dutch company that fully complies with the DCGC.
We intend to rely in certain cases on Nasdaq Stock Market rules that permit us to comply with applicable Dutch corporate governance practices, rather than the corresponding domestic U.S. corporate governance practices, and therefore our shareholders’ rights will differ from the rights they would have as a shareholder of a domestic U.S. issuer.
As a foreign private issuer whose ordinary shares are listed on the Nasdaq Capital Market, we are permitted in certain cases to follow Dutch corporate governance practices instead of the corresponding requirements of the Nasdaq Marketplace Rules. A foreign private issuer that elects to follow a home country practice instead of Nasdaq requirements must submit to Nasdaq in advance a written statement from an independent counsel in such issuer’s home country certifying that the issuer’s practices are not prohibited by the home country’s laws. In addition, a foreign private issuer must disclose in its annual reports filed with the SEC each such requirement that it does not follow and describe the home country practice followed instead of any such requirement. We intend to follow Dutch corporate governance practices with regard to the quorum requirements applicable to meetings of shareholders and the provision of proxy statements for general meetings of shareholders, rather than the corresponding domestic U.S. corporate governance practices. In accordance with Dutch law and generally accepted business practices, our articles of association do not provide quorum requirements (other than those which follow from Dutch law) generally applicable to general meetings of shareholders. Although we do intend to provide shareholders with an agenda and other relevant documents for the general meeting of shareholders, Dutch law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in the Netherlands. Accordingly, our shareholders may not be afforded the same protection as provided under Nasdaq’s corporate governance rules.
Any U.S. or other foreign judgments that investors may obtain against us may be difficult to enforce against us in the Netherlands.
We are incorporated under the laws of the Netherlands. We currently have only limited operations in the United States. Most of our assets are currently located in the Netherlands. The majority of our management board, supervisory board and officers reside outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce against them or the Company in U.S. courts, including judgments predicated upon the civil liability provisions of the federal securities laws of the United States. In addition, it is not clear whether a Dutch court would impose civil liability on the Company or any of our managing directors or supervisory directors in an original action based solely upon the federal securities laws of the United States brought in a court of competent jurisdiction in the Netherlands.
56
The United States and the Netherlands currently do not have a treaty providing for the reciprocal recognition and enforcement of judgments, other than arbitration awards, in civil and commercial matters. Consequently, a final judgment for payment given by a court in the United States, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in the Netherlands. In order to obtain a judgment which is enforceable in the Netherlands, the party in whose favor a final and conclusive judgment of the U.S. court has been rendered will be required to file its claim with a court of competent jurisdiction in the Netherlands. Such party may submit to the Dutch court the final judgment rendered by the U.S. court. If and to the extent that the Dutch court finds that the jurisdiction of the U.S. court has been based on grounds which are internationally acceptable and that proper legal procedures have been observed, the Dutch court will, in principle, give binding effect to the judgment of the U.S. court, unless such judgment contravenes principles of public policy of the Netherlands. Dutch courts may deny the recognition and enforcement of punitive damages or other awards. Moreover, a Dutch court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages. Enforcement and recognition of judgments of U.S. courts in the Netherlands are solely governed by the provisions of the Dutch Code of Civil Procedure.
Based on the lack of a treaty as described above, U.S. investors may not be able to enforce against us or our management board or supervisory board members, representatives or certain experts named herein who are residents of the Netherlands or countries other than the United States any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities laws.
Item 4: Information on the Company
A. History and Development of the Company
ProQR Therapeutics is a biotechnology company dedicated to changing lives by developing RNA therapies for severe rare and common diseases. We focus on advancing our proprietary Axiomer and Trident RNA-editing platform technologies.
ProQR was founded in 2012 by Daniel de Boer, Gerard Platenburg, the late Henri Termeer and Dinko Valerio. Since September 18, 2014, our ordinary shares have been listed on Nasdaq. They are currently trading on Nasdaq Capital Market under the ticker symbol “PRQR”. As of December 31, 2023, we had raised € 435 million in gross proceeds from our public offerings of shares and private placements of equity securities. In addition, we have received grants, loans and other funding from patient organizations, private lenders and government institutions supporting our programs, including from Foundation Fighting Blindness and the Dutch government under the innovation credit program.
Our legal name is ProQR Therapeutics N.V. and we were incorporated in the Netherlands on February 21, 2012. We reorganized from a private company with limited liability to a public company with limited liability on September 23, 2014. Our company is registered with the Dutch Trade Register of the Chamber of Commerce (‘handelsregister van de Kamer van Koophandel’) under number 54600790. Our corporate seat is in Leiden, the Netherlands. The address of our headquarters and registered office is Zernikedreef 9, 2333 CK Leiden, the Netherlands, telephone number +31 88 166 7000. Our U.S. office is located at 245 Main Street, Cambridge, MA 02142, USA. The name and address of our agent for service in the United States is Andrew Morris, 245 Main Street, Cambridge, MA 02142, USA.
The SEC maintains an internet site that contains reports, proxy and information statements, and other information regarding issuers like ProQR that file electronically with the SEC. The address of that website is www.sec.gov. We maintain a corporate website at www.ProQR.com. Information found on, or accessible through, our website is not incorporated by reference into and should not be considered a part of this Annual Report, and the reference to our website in this Annual Report is an inactive textual reference only.
57
B. Business Overview
We are a biotechnology company dedicated to the creation of transformative RNA therapies to improve the lives of patients and families affected by diseases with high unmet medical need. To achieve this, we are advancing our proprietary Axiomer RNA-editing platform technology. Our product candidates are designed to engage Adenosine Deaminase Acting on RNA (“ADAR”) to conduct targeted RNA editing which we believe have the potential to become a new class of innovative medicines with applicability to a broad range of therapeutic areas. Using our deep RNA expertise and our strong intellectual property position, we are advancing a platform to develop these RNA editing therapeutics, which we call “Editing Oligonucleotides”, or EONs, for a variety of human diseases.
Axiomer uses EONs to mediate single nucleotide changes to RNA in a highly specific and targeted way using molecular machinery that is present in human cells called ADAR. Axiomer EONs are designed to recruit and direct endogenously expressed ADARs to change an Adenosine (A) to an Inosine (I) in the RNA – an Inosine is translated as a Guanosine (G). This approach can be used to correct an RNA with a disease-causing mutation back to a normal (wild type) RNA, modulate protein expression, or alter a protein so that it will have a new function that helps prevent or treat disease.
Since discovering the Axiomer RNA editing technology in 2014, we have established a leading intellectual property estate in the ADAR editing space, defined the design ground rules, and optimized chemistries for therapeutic use.
Our research and development strategy focuses on the use of our Axiomer platform to develop novel RNA editing therapeutics to address diseases with high unmet medical need. We are initially focused on diseases originating in the liver and in the central nervous system (“CNS”) where research into human genetics has shown us that changing the RNA or correcting pathogenic mutations via A-to-I editing may lead to a benefit for patients. We prioritize areas with well-established biomarkers for the assessment of early clinical activity and to establish proof of target engagement, established clinically relevant endpoints, and the ability to leverage existing proven delivery technology. We are advancing AX-0810 for Cholestatic Diseases targeting Na-taurocholate cotransporting polypeptide, or NTCP, and AX-1412 for Cardiovascular Disease (“CVDs”) targeting Beta-1,4-galactosyltransferase 1, or B4GALT1, as our initial pipeline programs. In 2024, we announced a new research partnership with the Rett Syndrome Research Trust (“RSRT”) focused on utilizing Axiomer to develop EONs targeting an underlying genetic variant that causes Rett syndrome, a rare neurodevelopment disorder, which is included on our pipeline as AX-2402.
In addition to advancing our wholly-owned pipeline programs, we entered into a global licensing and research collaboration with Eli Lilly and Company in September 2021 where our Axiomer RNA editing platform is being used to progress new drug targets for disorders toward clinical development and commercialization. Initially focused on five targets, the partnership was expanded to ten targets in December 2022, with an option for further expansion to fifteen targets.
58
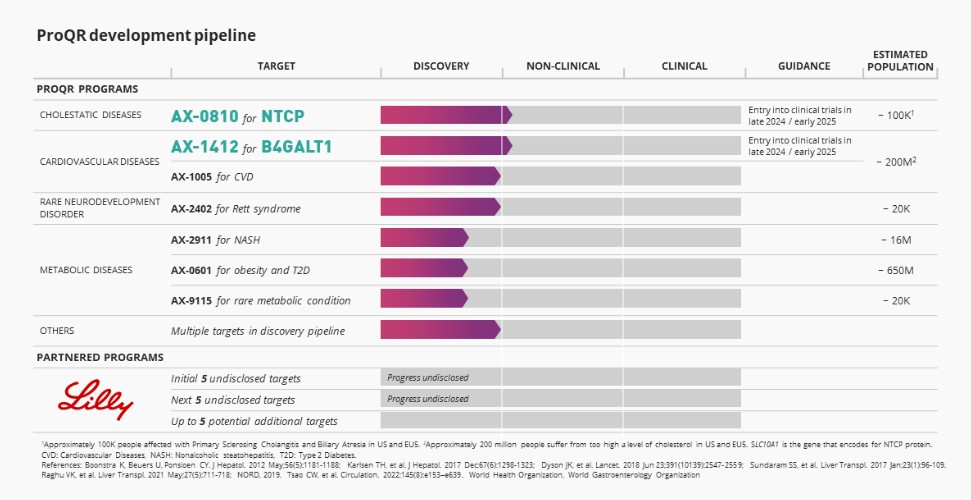
We believe the platform has significant potential to yield many additional therapeutic candidates. Thus, we continuously evaluate further opportunities for beneficial collaborations or strategic partnerships to efficiently advance product candidates with the goal of bringing medicines to patients.
We have other earlier stage RNA editing platform technologies, including our Trident platform. Our Trident RNA pseudouridylation platform is designed to enable the suppression of nonsense mutations and premature stop codons (“PTC”) that cause 11% of all human genetic diseases. Since all premature stop codons contain uridine, pseudouridylation of that uridine converts those nonsense codons into sense codons. The Trident technology harnesses the endogenously expressed pseudouridylation machinery with guide RNAs to, amongst other potential functions, inhibit nonsense messenger RNA (“mRNA”)-mediated decay (“NMD”) in a sequence-specific manner and promote PTC readthrough. The Trident technology has the potential to be applied in genetic diseases caused by PTCs.
Both the Axiomer and Trident platforms are novel, proprietary RNA editing technologies invented at ProQR or with our academic collaborators. We have built a broad intellectual property estate around these technologies and together with the leading academic experts in the RNA field, we continue to advance these technologies. RNA editing for therapeutic applications.
RNA antisense oligonucleotides (“AONs”) have been used as therapeutics for the last few decades. ProQR scientists have invented entirely new ways of using the proven modality of oligonucleotides to recruit a novel mechanism of action.
RNAs are produced in a process called transcription, where genetic information in DNA is copied into RNA. The information in RNA then serves as a blueprint to produce a protein via a process called translation. Before translation occurs, RNA can be processed in several ways. One way is RNA editing, which involves changing specific nucleotides, or letters, in the RNA code. RNA editing is a naturally occurring process that helps ensure that produced proteins function normally. It can also create slightly differently functioning proteins.
59
One common type of RNA editing is A-to-I editing, where Adenosines (abbreviated as “A”), are changed into Inosines (abbreviated as I), as shown in Figure 1. Nucleotides pair together to create double stranded structures within the RNA. Double stranded RNA structures are found and bound to by ADAR, which is naturally present in the cells. ADAR then can edit As into Is, which is read by a ribosome as a G, or guanosine. This process is called “A to I” editing, which functionally enables changing an A into a G. In 2014, scientists at ProQR invented Axiomer, which was conceived based on the idea of recruiting endogenous ADAR in humans to make single A to I changes in RNA in a highly specific and targeted manner, using EONs as shown in Figure 1b.
Figure 1a (left): RNA editing is a naturally occurring process whereby ADARs perform A to I editing.
Figure 1b (right): ProQR’s Axiomer RNA editing technology platform uses EONs to recruit and direct endogenously expressed ADARs to edit an A to an I in the RNA, which is then translated as a G, allowing highly specific editing.
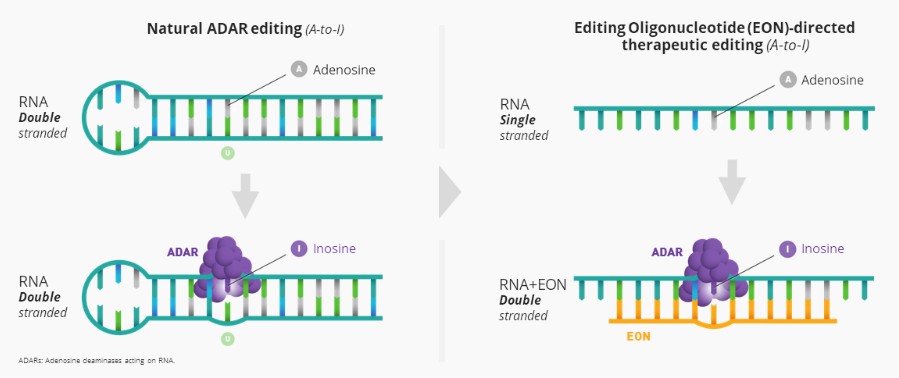
There are over 16 million known locations in the RNA where ADARs perform A to I editing throughout the body, which we believe represents a powerful potential therapeutic mechanism for multiple disease areas. Axiomer could potentially yield a new class of medicines for both rare and prevalent diseases with unmet need.
Our Strategy
We are advancing Axiomer as a platform to develop a new class of innovative medicines based on ADAR RNA editing, which we believe has the potential to treat a broad range of diseases that currently lack adequate treatment options. Our novel and proprietary RNA editing platform technologies, known as Axiomer and Trident, are new ways to use oligonucleotides to edit single nucleotides in the RNA. We believe the Axiomer technology may be applicable to thousands of disease-causing mutations by correcting RNA in genetic diseases. Beyond mutation correction, Axiomer also has the potential to address unmet medical needs in common conditions, by modulating protein expression or altering a protein so that it will have a new function to help prevent or treat diseases. We intend to continue to optimize our platform as we advance to clinical stage and beyond. Key elements of our strategy include:
| ● | Pipeline: We intend to use these platforms to develop novel therapies initially for targets related to liver- and CNS-originating diseases, and beyond. With our Axiomer RNA-editing technology platform, we are advancing AX-0810 for Cholestatic Diseases targeting NTCP and AX-1412 for CVDs targeting B4GALT1 as our initial pipeline programs. In January 2024, we announced a partnership with the RSRT in which we will develop EONs targeting an underlying genetic variant that causes Rett syndrome, a rare neurodevelopment disorder, which is included on our pipeline as AX-2402. |
60
| ● | Partnerships: We continue to validate and create value for these platforms by selectively pursuing additional licensing, partnering, and other strategic relationships outside of our core focus area, such as our partnership with Lilly, and the RSRT. |
We seek to maximize the value of our pipeline by retaining development and commercialization rights to those product candidates, indications and geographies that we believe we can independently develop, seek approval for, and commercialize on our own. Beyond this, for other product candidates, such as those for more prevalent indications, and other geographies, we plan to selectively and opportunistically seek potential partnerships following early-stage clinical proof of concept.
Our Novel Axiomer RNA Editing Technology Platform
Antisense oligonucleotides, or AONs, have been used as therapeutics for decades. Our Axiomer RNA editing technology is based on oligonucleotides that are called editing oligonucleotides, or EONs, designed to recruit endogenous ADAR enzymes (“Adenosine Deaminases Acting on RNA”) to make single adenosine-to-inosine (A-to-I) changes in the RNA in a highly specific and targeted manner. This technology could correct thousands of G-to-A mutations in the human population that cause diseases. In vitro and in vivo work indicates that the EONs are generally applicable for the correction of RNA G-to-A mutations. The technology is also designed to modulate protein expression or alter proteins to provide a new function to help prevent or treat disease. With this applicability, we believe Axiomer has the potential to address hundreds of genetic and non-genetic diseases.
Across a range of targets, we have shown both in vitro and in vivo platform proof-of-concept for our Axiomer RNA editing technology platform, in cell models, organoids, and animal models, including relevant higher order species.
Our Axiomer platform has demonstrated proof of concept across multiple models, as shown in Figure 2a and 2b reporting up to 70% editing of ACTB in the liver of non-human primates (“NHPs”) and mice and 50% editing in the nervous system.
Figure 2a: Robust editing with Axiomer editing oligonucleotides reported in the nervous system across different models and targets in vivo including non-human primates.
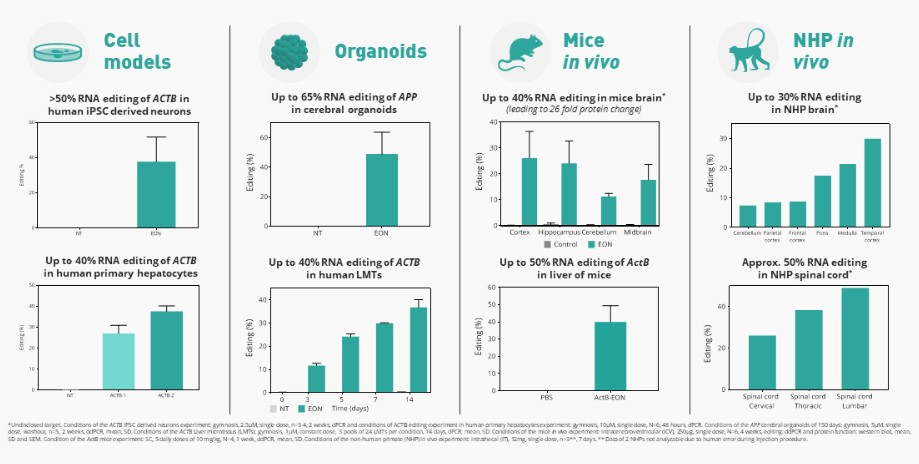
61
Figure 2b: Up to 70% editing of ACTB in the liver of non-human primates (“NHPs”) and mice.
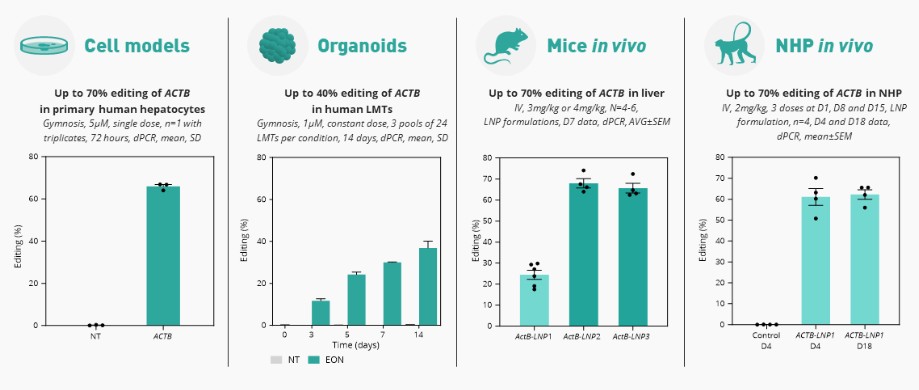
Additionally, across EONs, preliminary nonclinical safety assessment showed a similar safety profile compared to other single-stranded RNA oligonucleotides.
If translated in human testing, we believe the editing activity and safety profile supports the potential of our technology and plan to advance product candidates based on our Axiomer platform to clinical stage.
Our Pipeline Programs
We are advancing Axiomer as a platform to develop a new class of innovative medicines based on RNA editing. Our initial pipeline targets focus on liver-originating and CNS diseases share several key characteristics, including:
| ● | Population with unmet need |
| ● | Target with deep roots in human genetics |
| ● | Preclinical models with strong translatability into the clinic |
| ● | Validated biomarkers to assess target engagement and the ability to have early insight into safety |
| ● | Established disease-specific clinical endpoints |
Our initial pipeline programs include AX-0810 for Cholestatic Diseases targeting NTCP and AX-1412 for CVDs targeting B4GALT1. During 2024, we plan to present non-clinical proof-of-concept data for these programs and anticipate providing an update on translational data to enable submission of Clinical Trial Application (“CTA”). We expect to advance these programs to clinical trials in late 2024 / early 2025.
In January 2024, we announced a partnership with the RSRT in which we will develop EONs targeting an underlying genetic variant that causes Rett syndrome, a rare neurodevelopment disorder, which is included on our pipeline as AX-2402.
62
AX-0810 for Cholestatic Diseases targeting NTCP
Cholestatic Diseases overview
Cholestatic Diseases are caused by a toxic buildup of bile acids in the liver due to bile duct dysfunction, which causes liver cell damage. The consequences of these disorders can be devastating and significantly impact a person's quality of life, including pruritus, dry skin, fatigue, pain, weight loss, and many others. Without treatment, the damage progresses through various stages, from fibrosis to cirrhosis, ultimately leading to liver failure and an increased risk of liver cancer. Liver transplants are often necessary for primary sclerosing cholangitis (“PSC”) and biliary atresia (“BA”), two forms of Cholestatic Diseases with high unmet medical needs.
PSC is a condition that causes inflammation and is typically diagnosed in people aged 30 to 40, more commonly affecting men (66%). It is estimated that 80,000 people in North America and Europe have PSC, with a prevalence of 1 to 9 individuals per 100,000. This condition causes fibrosis and sclerosis of bile ducts, leading to a toxic buildup of bile acids in the liver.
BA is a pediatric condition that affects newborns, resulting from the absence or defect of bile ducts. This condition causes harmful bile acids to accumulate in the liver, leading to rapid progression to cirrhosis early in life. It is estimated that 20,000 individuals in North America and Europe have BA, with a prevalence of 1 in 10,000 to 15,000 births in the western world.
Limitations of the Current Treatment Landscape
Currently, there are no approved drugs for treating PSC and BA. For PSC, liver transplantation is the only treatment option with evidence to extend survival. However, PSC can return in 20 to 40% of patients who undergo liver transplantation, and the median survival without a transplant is only 21 years. Surgery in the first weeks of life for BA is the gold standard treatment. However, most patients who receive this surgery will still require a liver transplant early in life.
AX-0810 for Cholestatic Diseases targeting NTCP
The liver cells mainly obtain bile acids from the enterohepatic reuptake cycle. The process is primarily carried out by a transporter called Na-taurocholate transporting polypeptide (“NTCP, SLC10A1”), which takes bile acids from the portal circulation to the liver. Studies show that inhibiting NTCP can improve liver function by reducing the levels of toxic bile acids, improving liver damage markers (fibrosis, cholangiocyte proliferation, Alkaline phosphatase or ALP, alanine transaminase or ALT), and lowering inflammation biomarkers (“cytokines”).
AX-0810, our Axiomer-targeted RNA editing oligonucleotide, aims to reduce the reuptake of bile acids in the liver by inhibiting NTCP function. Variants in NTCP that change its capacity to recycle bile acid into the liver naturally occur in some people without causing any symptoms associated with cholestasis. This finding suggests that our approach is safe and may reduce the accumulation of toxic bile acids in the liver. Moreover, such variants in NTCP also promote the elimination of bile acids from the body by increasing their excretion in the feces and urine, a process called sulfation of bile acids, which enhances their solubility and reduces their absorption in the intestines. Based on its mechanism of action, we believe AX-0810 may have the potential to modify the course of Cholestatic Diseases, delay or prevent complications such as cirrhosis and liver failure, and alleviate associated symptoms.
AX-1412 for Cardiovascular Disease targeting B4GALT1
Cardiovascular Disease overview
Cardiovascular Diseases (“CVDs”) are a group of health conditions that affect the heart and blood vessels, such as atherosclerosis which can lead to severe problems like heart attacks, heart failure, and stroke. The World Health Organization (“WHO”) has identified unhealthy diet, physical inactivity, tobacco use, and excessive alcohol consumption as major behavioral risk factors for heart disease and stroke, increasing intermediate risk factors including but not limited to high blood pressure, cholesterol, glucose levels, and obesity.
63
CVDs are the leading cause of disability and death globally, becoming a significant health issue worldwide. Approximately 18 million people die from CVDs each year, making up 32% of all global deaths, according to a report by the World Health Organization in 2021. In the United States, the American Heart Association estimates that by 2035, more than 130 million adults will have some form of CVD.
Current Treatment Landscape and Limitations
CVD treatment involves taking medications to lower cholesterol and blood pressure levels. The most common drugs are statins, ezetimibe, and PCSK9 inhibitors. These medications are primarily used to lower LDL cholesterol levels. Other treatments, such as ANGPTL3 inhibitors, decrease the residual risk of heart disease in patients with high LDL cholesterol levels. However, even with these therapies, less than 35% of Americans with high LDL cholesterol levels reach their target levels recommended by guidelines. CVD events still occur even when LDL cholesterol levels meet clinical goals. Many patients also struggle to continue taking their medications long-term, with less than 50% of patients taking their LDL-lowering medicines 2 years after a CVD event. Additionally, 5 to 10% of patients cannot tolerate high doses of statins, primarily due to muscle aches.
AX-1412 for Cardiovascular Disease targeting B4GALT1
AX-1412 represents a potential targeted approach to RNA editing of B4GALT1 that leads to a promising strategy for protecting against CVDs by simultaneously lowering levels of LDL-c and fibrinogen. Recent gene-based analysis has shown that rare protective variants changing protein activity and predicted deleterious missense variants in B4GALT1 are associated with a decreased risk of coronary artery disease. Additionally, a particular missense variant (p.Asn352Ser) in the beta-1,4-galactosyltransferase 1 B4GALT1 gene is prevalent in the Amish population and associated with lower levels of LDL-c and CVDs.
The beneficial effects of these genetic variations are due to the hypo-galactosylation of apolipoprotein B100 and fibrinogen, which are known to be independent drivers of an increased risk of CVDs, as well as immunoglobulin G and transferrin. However, it's important to note that studies have shown that B4GALT1 knockdown can lead to semi-lethality and severe developmental abnormalities in mice models and therefore we believe B4GALT1 inhibition is not a feasible therapeutic approach for this purpose.
Although there are several approaches to lowering the risks of CVDs, including reducing LDL-c and ApoB levels, reducing fibrinogen levels may offer additional benefits to patients with unmet medical needs in this large population. Fibrinogen reduction can be used either as a stand-alone therapy or an adjunct therapy to other treatments.
We are developing Axiomer targeted RNA EON AX-1412 to address CVD by editing B4GALT1. RNA editing to a protective variant of B4GALT1 can have positive effect on CVDs risk factors by leading to hypo-galactosylation of apolipoprotein B100 and fibrinogen. Based on its mechanism of action, we believe that AX-1412 is a novel and unique approach to address CVD by lowering LDL-C and fibrinogen levels ultimately leading to a reduced residual risk in CVDs.
We intend to advance AX-1412 targeting B4GALT1 to early clinical proof of concept stage, then would seek to partner this program.
64
Our Earlier-Stage/Discovery Programs
In January 2024, we announced a partnership with the RSRT that will focus on the design and development of EONs using our Axiomer technology platform targeting the transcription factor MECP2 and correcting mutations of interest. AX-2402 is our program focusing on Rett syndrome, which is a progressive neurodevelopmental disorder caused by genetic mutations in the Methyl CpG binding protein 2 (“MECP2”) and diagnosed primarily in females. It is characterized by apparently normal psychomotor development during the first six to 18 months after birth, followed by a period of developmental stagnation, then a regression in language and motor skills, followed by long-term relative stability. During the phase of regression, affected patients develop repetitive, stereotypic hand movements that replace purposeful hand use. Additional symptoms include gait ataxia and apraxia, seizures, tremors, episodic apnea and/or hyperpnea, gastrointestinal issues, scoliosis and musculoskeletal problems, anxiety and sleep issues and bruxism.
In addition to AX-2402 for Rett syndrome, we have multiple other early-stage research programs ongoing that target additional diseases with our Axiomer EON approach, including AX-1005 for undisclosed targets in CVD, AX-2911 for nonalcoholic steatohepatitis (“NASH”), AX-0601 for obesity and Type 2 diabetes, AX-9115 for rare metabolic condition, as well as multiple other targets in our discovery pipeline.
Our Partnership Strategy
Our business strategy is to develop and ultimately commercialize a broad pipeline of RNA therapies based on our Axiomer RNA editing platform technology. We are initially focused on developing an internal pipeline based on liver-originating diseases, including Cholestatic Diseases and CVD, among others. We believe there is broad applicability of the platform beyond liver and as part of the strategy to advance Axiomer, we have entered into, and expect to enter into additional collaboration and licencing agreements as a means of obtaining funding and capabilities to advance programs based on Axiomer.
A global licensing and research collaboration with Eli Lilly and Company focuses on the discovery, development, and commercialization of potential new medicines for genetic disorders using our Axiomer RNA editing technology with a focus on CNS and peripheral nervous system, (“PNS”). The partnership, formed in 2021, initially focused on up to five targets. In December 2022, the partnership was expanded to up to ten targets, with an option for an additional five targets. Under the terms of the agreements, we received $ 125 million upfront from Lilly and would be paid an additional $ 50 million if Lilly exercises the option for five additional targets. We are also eligible to receive up to approximately $ 3.75 billion in milestones, as well as royalties on potential product sales.
In January 2024, we announced a collaboration with the RSRT, as described above. RSRT awarded ProQR approximately $ 1 million as a research grant for the initial phase of the project, which will encompass editing oligonucleotide design and optimization, including evaluation in in vivo models for editing efficacy and MECP2 protein recovery. It is the intent of the partnership to be continued by an expanded co-funding arrangement following the initial discovery work. The co-funding of the next phase of the collaboration would enable clinical development of an Axiomer-based therapeutic for Rett syndrome MECP2.
We believe the platform holds significant further potential for strategic transactions.
Ophthalmology Assets
In August 2022, we made the decision to exclusively focus our strategy on the advancement of our Axiomer RNA editing technology and to partner our ophthalmology programs. In December 2023, we announced that we had completed a transaction divesting the late stage ophthalmic assets sepofarsen and ultevursen to Laboratoires Théa (“Théa”) who will continue the development of these therapies for patients with LCA10 and Usher Syndrome. Under the terms of the agreement, ProQR received an initial payment of € 8 million and may be eligible for up to € 165 million in further development, regulatory, and commercial earn-out payments upon related achieved milestones, as well as double-digit royalties based on commercial sales in the United States and EU.
65
Competition
The pharmaceutical industry is highly competitive and subject to rapid and significant technological change. Our potential competitors include large pharmaceutical, biotechnology, specialty pharmaceutical, and generic drug companies, academic institutions, government agencies and research institutions. Key competitive factors affecting the commercial success of our product candidates are likely to be efficacy, safety and tolerability profile, delivery, reliability, convenience of dosing, patient recruitment for clinical studies, price and reimbursement. Many of our existing or potential competitors have substantially greater financial, technical, and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA, EMA and other regulatory approvals of products and the commercialization of those products. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a small number of our competitors. Accordingly, our competitors may be more successful than we may be in obtaining FDA or EMA approval for therapies and achieving widespread market acceptance. Our competitors’ products may be more effective, or more effectively marketed and sold, than any product we may commercialize and may render our therapies obsolete or non-competitive before we can recover development and commercialization expenses.
Our competitors are working on similar technologies in the field of RNA editing, but also in the field of gene editing and gene therapy as well as other types of therapies, such as small molecules, protein replacement or antibodies.
Human Capital Resources
We believe in passion and commitment and have built a strong team of ProQRians from all walks of life and over 25 different nationalities, who are up to the challenge and committed to make a difference for the patients we serve. We actively create a caring atmosphere, in which we love to work and maintain productive and happy lives. At ProQR, we foster empowerment, self-development, creativity, and a sense of community.
As an employer, we are a true believer in the value of a workforce in which people from all backgrounds are encouraged to develop themselves both personally and professionally. This is reflected in our equal gender balanced leadership team and broader workforce. We believe that happy and energized people, working well together in an environment in which they thrive, will do phenomenal and awesome things.
We are committed to ensure that no employee, candidate, or job applicant receives less favorable treatment on the grounds of race, age, disability, pregnancy, religion, gender identity and expression, sexual orientation, marriage or civil partnership status. At ProQR, we want to create an inclusive culture where everyone can be valued for who they are and in which individual differences and the contributions in all forms are recognized and valued.
Animal Welfare
It is required by regulatory authorities to demonstrate the safety and, if possible, efficacy of a new drug in animals before it can be tested in humans. The welfare of animals in our preclinical studies is of great importance to us for reasons of ethics, quality, reliability, and applicability of scientific studies. To assure high quality (scientific) research, animal welfare is essential. By actively pursuing the 3R principles (Reduce, Refine and Replace), ProQR is committed to reduce the number of animals needed, minimize discomfort and pain of animals used, and use alternatives to animal research whenever possible.
Animal experiments will be performed only if there are no alternatives such as performing in silico, in vitro or ex vivo studies. Study designs will be evaluated with the aim to identify opportunities to reduce the number of animals needed to achieve the objectives of the study. By conducting small pilot (tolerability) studies and by using innovative new technologies and modeling approaches, we further pursues the ambition to reduce, refine and replace animal studies.
Approval by the (institutional or national) animal care and use committees is required prior the execution of in vivo studies.
66
External collaborators contracted for the execution of our in vivo preclinical studies, also known as contract research organizations (“CROs”), are selected based on their expertise, quality and accreditations for laboratory animal care and welfare. CRO facilities are audited by us prior to contracting to ensure that the housing, husbandry and welfare of animals complies with the highest international standards. Personnel responsible for housing, husbandry and the care of animals must have received adequate and relevant documented education.
Manufacturing and Supply
ProQR does not own or operate GxP manufacturing facilities to produce clinical or commercial drug product. Historically manufacture of drug substance and drug product for clinical use was outsourced to approved vendors under service agreements.
Currently, manufacture of small scale non GxP drug substance and drug product batches to support the discovery and initial pre-clinical phase of the Axiomer platform are manufactured in our in-house laboratories. As the portfolio progresses, larger scale batches and those intended for clinical use will be manufactured at approved vendors under phase appropriate service agreements.
Our vendor management strategy will ensure that we have sufficient capacity to meet the demands of the portfolio now and in the future. The contract manufacturing organizations selected to manufacture our product candidates under cGMP conditions are assessed and audited to ensure that they meet the extensive regulations imposed e.g. technical expertise, facility and equipment management, documentation, data integrity, manufacturing processes and controls, personnel, quality control and quality assurance.
Intellectual Property
We strive to protect our technology platforms and our product candidates through a variety of approaches, including obtaining and maintaining patent and trade secret protection for our current and future technology platforms, our product candidates as well as their methods of use and processes for their manufacture, and any other inventions or discoveries that are commercially important to our business. We seek to obtain domestic and international patent protection and endeavor to promptly file patent applications or in-license patent rights relating to new, commercially valuable inventions to expand our intellectual property portfolio.
Our commercial success will depend in part upon obtaining and maintaining patent and trade secret protection for our current and future technology platforms and product candidates, as well as successfully defending our patent rights against third-party challenges. Our ability to prevent or stop third parties from making, using, selling, offering to sell or importing our product candidates will depend in part upon whether we have valid and enforceable patent rights that cover the activities of third parties.
We cannot be sure that patents will be granted with respect to any of our own or our in-licensed pending patent applications or with respect to any patents applications we may own or in-license in the future, nor can we be sure that any of our own or in-licensed patents or any patents we may own or license in the future will be useful in protecting our technology. Please see “Risk Factors—Risks Related to Our Intellectual Property” for additional information on the risks associated with our intellectual property strategy and portfolio.
Patent Rights
We have been building and will continue to build our patent portfolio. Where possible, we pursue or plan to pursue multi-tiered patent protection for our technology platforms and our product candidates as well as their manufacture, formulation and use. In addition to the United States, we file, and plan to file, patent applications in various countries and regions where we think such filings are likely to be cost effective.
67
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file or plan to file, the patent term is 20 years from the earliest date of filing of a non-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. PTO in granting a patent. However, the term of a United States patent may be shortened if a patent is terminally disclaimed by its owner over another patent. In addition, depending upon the circumstances, certain United States patents may be eligible for a patent term extension which compensates a patentee for delays in granting marketing approval for a patented active ingredient or use of an active ingredient. In Europe, a similar mechanism is available, such that patents may be eligible for a supplementary protection certificate to compensate for the time lost in obtaining marketing authorization for the active ingredient.
Patent Rights Relating to Our Axiomer Program
ProQR’s Axiomer RNA editing technology platform uses EONs to recruit and direct endogenously expressed ADARs to edit an A to an I in the RNA, which is then translated as a G, allowing highly specific editing. Since 2014, when the first inventions were conceived at ProQR, we have been filing patent applications for intellectual property rights related to our Axiomer platform. Many of these claim EONs with specific features that allow them to guide recruitment of endogenous ADAR for the purpose of therapeutic RNA editing, without the need of ADAR overexpression or artificial ADAR recruitment systems. Further to that, we have grown a strong intellectual property position for EONs that can bring about RNA editing in RNA to yield a gain-of-function alteration or a loss-of-function alteration in a wide variety of therapeutic areas. We rely, and continue to rely, on intellectual property rights that are fully owned by us, or that have been co-filed with our research collaborators.
With regard to our Axiomer program, we filed the following international patent applications from 2015 to 20, several of which were continued in national and regional patent applications after the respective international phases.
PCT/EP2015/080347 – Granted in Brazil (BR 112017011510-7), Canada (CA 2,968,336), China (ZL 201580069286.1), Europe (EP 3234134 B1), Israel (IL 252386), India (IN 452659), Japan (JP 6718872), New Zealand (NZ 732182), Russia (RU 2711506), South Africa (ZA 2017/03464) and the U.S. (US 10,676,737; US 11,781,134). Pending in Australia and the U.S. (continuation application). The term of any patents resulting from these applications would be expected to extend to at least 2035.
PCT/EP2017/065467 – Granted in Australia (AU 2017281497), Israel (IL 263332), Japan (JP 7074345), South Korea (KR 10-2418185) and the U.S. (US 10,988,763; US 11,649,454). Pending in Canada, China, Eurasia, Europe, New Zealand, and the U.S. (allowed continuation application). The term of any patents resulting from these applications would be expected to extend to at least between 2037.
PCT/EP2017/071912 – Granted in China (CN 110352244 B), Europe (EP 3507366 B1), Japan (JP 2019-511856), New Zealand (NZ 751483), South Korea (KR 10-2501980), South Africa (ZA 2019/01016) and the U.S. (US 10,941,402; US 11,851,656). Allowed in Australia. Pending in Canada, Israel, India, and the U.S. (continuation application). The term of any patents resulting from these applications would be expected to extend to at least between 2037.
PCT/EP2018/051202 – Granted in the U.S. (US 11,274,300). Pending in Europe. The term of a patent resulting from this application would be expected to extend to at least between 2038.
PCT/EP2019/062163 – Pending in Australia, Canada, Europe, Japan, New Zealand, and the U.S. The term of any patents resulting from these applications, if issued, would be expected to extend to at least 2039.
PCT/EP2020/053283 – Pending in Australia, Canada, Europe, Israel, New Zealand, and the U.S. The term of any patents resulting from these applications, if issued, would be expected to extend to at least 2040.
PCT/EP2020/059369 – Pending in Australia, Canada, China, Europe, Israel, Japan, New Zealand, and the U.S. The term of any patents resulting from these applications, if issued, would be expected to extend to at least 2040.
PCT/EP2020/060291 – Pending in Australia, Canada, Europe, Israel, New Zealand, and the U.S. The term of any patents resulting from these applications, if issued, would be expected to extend to at least 2040.
68
PCT/US2020/037580 – Granted in South Africa (ZA 2021/09497). Pending in Australia, Canada, China, Europe, Israel, India, Japan, New Zealand, and the U.S. We filed this application together with The Regents of the University of California as a co-applicant. In the Axiomer program we are working together with Dr. Peter Beal of the University of California, Davis, CA, USA. The term of any patents resulting from these applications, if issued, would be expected to extend to at least 2040.
PCT/EP2021/070535 – Pending in Australia, Canada, Europe, Japan, and the U.S. The term of any patents resulting from these applications, if issued, would be expected to extend to at least 2041.
PCT/EP2023/053503 – PCT pending. The term of any patents resulting from this application, if issued, would be expected to extend to at least 2043.
PCT/EP2023/069612 – PCT pending. We filed this application together with The Regents of the University of California as a co-applicant. In the Axiomer program we are working together with Dr. Peter Beal of the University of California, Davis, CA, USA. The term of any patents resulting from this application, if issued, would be expected to extend to at least 2043.
PCT/EP2023/069609 – PCT pending. We filed this application together with The Regents of the University of California as a co-applicant. In the Axiomer program we are working together with Dr. Peter Beal of the University of California, Davis, CA, USA. The term of any patents resulting from this application, if issued, would be expected to extend to at least 2043.
Patent Rights Relating to Our Trident Program
With regard to our TRIDENT program, we filed an international patent application (PCT/US2019/024282) in 2019 directed to antisense oligonucleotides that are applicable for nucleotide-specific pseudouridylation. A patent was granted in South Africa (ZA 2020/05217) and in Europe (EP 3775210 B1). The US application was allowed, and applications are currently pending in Australia, Canada, China, Europe (divisional application), Israel, India, Japan, New Zealand, and the U.S. (continuation application). In the TRIDENT program we are working together with Prof. Yi-Tao Yu at the University of Rochester, NY, USA. The term of any patents resulting from these applications would be expected to extend to at least 2039.
Trade Secrets
In addition to patent protection, we rely on trade secrets and know-how to develop and maintain our competitive position. Trade secrets and know-how can be difficult to protect. Nevertheless, we seek to protect our trade secrets and know-how via, among other things, confidentiality and invention assignment agreements with our employees and consultants. We also seek to preserve the confidentiality of our trade secrets and know-how by implementing and maintaining security of our premises and information and limiting access to our trade secrets and know-how.
License Agreements
In February 2019, we entered into an agreement with the University of Rochester, New York, which gives us a world-wide, exclusive, royalty-bearing, sublicensable license in the field of antisense oligonucleotides for use in nucleotide specific RNA editing through pseudouridylation, under certain patent rights of University of Rochester. This license agreement contains certain diligence obligations for the Company coupled to milestone payments and complements the Company’s intellectual property relating to the Trident program. The royalties payable under this license agreement are in the low single digits.
69
Regulatory Matters
Government Regulation and Product Approval
Government authorities in the United States at the federal, state and local level, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing, export and import of products such as those we are developing.
U.S. Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations and other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices, or GLP, regulations; |
| ● | manufacture of the drug product in accordance with current Good Manufacturing Practice, or cGMP, regulations; |
| ● | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| ● | performance of adequate and well-controlled human clinical trials according to Good Clinical Practice, or GCP, regulations to establish the safety and efficacy of the proposed drug for its intended use; |
| ● | preparation and submission to the FDA of a new drug application, or NDA; |
| ● | payment of user fees for FDA review of an NDA; |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP; |
| ● | satisfactory completion of an FDA audit of clinical trial sites to assess compliance with GCP; and |
| ● | review and approval by the FDA of the NDA. |
The testing and approval process require substantial time, effort and financial resources and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
70
Once a pharmaceutical product candidate is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND. The sponsor must also include a protocol detailing, among other things, the objectives of the initial clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions related to a proposed clinical trial and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during clinical trials due to safety concerns or non-compliance and may be imposed on all drug products within a certain class of drugs. The FDA also can impose partial clinical holds, for example, prohibiting the initiation of clinical trials of a certain duration or for a certain dose.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent in writing before their participation in any clinical trial. Further, an IRB must review and approve the plan for any clinical trial before it commences at any institution, and the IRB must conduct continuing review and reauthorize the trial at least annually. An IRB considers, among other things, whether the risks to individuals participating in the clinical trial are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Each new clinical protocol and any amendments to the protocol must be submitted for FDA review, and to the IRBs for approval. Protocols detail, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, the parameters to assess efficacy and the parameters to be used to monitor subject safety.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
Phase 1. The product is initially introduced into a small number of healthy human subjects or patients and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion and, if possible, to gain early evidence on effectiveness. In the case of some products for severe or life-threatening diseases, especially when the product is suspected or known to be unavoidably toxic, the initial human testing may be conducted in patients.
Phase 2. Involves clinical trials in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage and schedule.
Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population. These clinical trials are intended to establish the overall risk/benefit relationship of the product and provide an adequate basis for product labeling.
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
71
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA. Within 15 calendar days after the sponsor determines that the information qualifies for reporting, written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected suspected adverse events, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator's brochure, or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the product drug. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether a trial may move forward at designated checkpoints based on access to certain data from the trial.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
A drug being studied in clinical trials may be made available to individual patients in certain circumstances outside of clinical trials, known as expanded access or “compassionate use”. There is no requirement for a company to provide expanded access to its investigational product. However, if a company decides to make its investigational product available for expanded access, FDA reviews each request for expanded access and determines if treatment may proceed. Under the FDCA, a sponsor of one or more investigational products for the treatment of a serious disease or condition is required to make publicly available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational drug. This requirement applies on the earlier of the first initiation of a Phase 2 or Phase 3 trial of the investigational drug, or as applicable, 15 days after the drug receives a designation as a breakthrough therapy, fast track product, or regenerative advanced therapy.
U.S. Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the drug, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA for a new drug, requesting approval to market the product. The submission of an NDA is subject to the payment of a substantial user fee; a waiver of such fee may be obtained under certain limited circumstances. For example, the agency will waive the application fee for the first human drug application that a small business or its affiliate submits for review. The holder of an approved NDA is also subject to an annual prescription drug product program fee.
72
The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be re-submitted with the additional information. The re-submitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant to assure the product’s identity, strength, quality and purity. Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is or will be manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. An advisory committee is a panel of experts, including clinicians and other scientific experts, who provide advice and recommendations when requested by the FDA. The FDA is not bound by the recommendation of an advisory committee, but it considers such recommendations when making decisions.
The approval process is lengthy and difficult, and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA in its present form. The complete response letter usually describes all of the specific deficiencies that the FDA identified in the NDA that must be satisfactorily addressed before it can be approved. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application or request an opportunity for a hearing.
If a product receives regulatory approval, the approval may be significantly limited to specific patients and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require post-approval studies, including Phase 4 clinical trials, to further assess a drug’s safety and effectiveness after NDA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA may require a Risk Evaluation and Mitigation Strategy, or REMS, program as a condition of approval, which could entail requirements for long-term patient follow-up, a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
Fast Track Designation and Accelerated Approval
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life-threatening disease or condition for which there is an unmet medical need and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug may request the FDA to designate the drug as a Fast Track product concurrently with, or at any time after, submission of an IND, and the FDA must determine if the drug candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request. In addition to other benefits, such as the ability to engage in more frequent interactions with the FDA, the FDA may initiate review of sections of a Fast Track drug’s NDA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of each portion of the NDA and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted. Additionally, the Fast Track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
73
Under FDA’s accelerated approval regulations, the FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. In clinical trials, a surrogate endpoint is a marker, such as a measurement of laboratory or clinical signs of a disease or condition that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of post-approval clinical trials sometimes referred to as Phase 4 trials to confirm the effect on the clinical endpoint. Further, under the Food and Drug Omnibus Reform Act of 2022, or FDORA, the FDA may require, as appropriate, that such trials be underway prior to approval or within a specific time period after the date of approval for a product granted accelerated approval. Under FDORA, the FDA has increased authority for expedited procedures to withdraw approval of a drug or indication approved under accelerated approval for failure to conduct required post-approval studies with diligence, or to confirm a clinical benefit during post-marketing studies. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
Breakthrough Designation
A drug product can be designated as a breakthrough therapy if it is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that it may demonstrate substantial improvement over available therapies on one or more clinically significant endpoints. A sponsor may request that a drug product be designated as a breakthrough therapy concurrently with, or at any time after, the submission of an IND, and the FDA must determine if the drug candidate qualifies for breakthrough therapy designation within 60 days of receipt of the sponsor’s request. If so designated, the FDA shall act to expedite the development and review of the product’s marketing application, including by meeting with the sponsor throughout the product’s development, providing timely advice to the sponsor to ensure that the development program to gather preclinical and clinical data is as efficient as practicable, involving senior managers and experienced review staff in a cross-disciplinary review, assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor, and taking steps to ensure that the design of the clinical trials is as efficient as practicable.
Platform Technology Designation
Under FDORA, a platform technology incorporated within or utilized by a drug or biological product is eligible for designation as a designated platform technology if (1) the platform technology is incorporated in, or utilized by, a drug approved under an NDA or biologics license application, or BLA; (2) preliminary evidence submitted by the sponsor of the approved or licensed drug, or a sponsor that has been granted a right of reference to data submitted in the application for such drug, demonstrates that the platform technology has the potential to be incorporated in, or utilized by, more than one drug without an adverse effect on quality, manufacturing, or safety; and (3) data or information submitted by the applicable person indicates that incorporation or utilization of the platform technology has a reasonable likelihood to bring significant efficiencies to the drug development or manufacturing process and to the review process. A sponsor may request the FDA to designate a platform technology as a designated platform technology concurrently with, or at any time after, submission of an IND for a drug that incorporates or utilizes the platform technology that is the subject of the request. If so designated, the FDA may expedite the development and review of any subsequent original NDA or BLA for a drug that uses or incorporates the platform technology. Designated platform technology status does not ensure that a drug will be developed more quickly or receive FDA approval. In addition, the FDA may revoke a designation if the FDA determines that a designated platform technology no longer meets the criteria for such designation.
74
Priority Review
The FDA may grant an NDA a priority review designation, which sets the target date for FDA action on the application for a new molecular entity at six months after the FDA accepts the application for filing. Priority review is granted where there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition compared to available therapies. If criteria are not met for priority review, the application is subject to the standard FDA review period of ten months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Rare Pediatric Disease Priority Review Voucher
The FDA may grant rare pediatric disease designation for indications in the treatment or prevention of a rare disease or condition that affects fewer than 200,000 individuals in the United States and that is a serious or life-threatening disease that primarily affects individuals aged from birth to 18 years, including age groups often called neonates, infants, children and adolescents. Under the FDCA, a sponsor who receives approval of an NDA for a product that is for the prevention or treatment of a rare pediatric disease and meets certain additional criteria, may qualify for a rare pediatric disease priority review voucher, or PRV. A PRV can be redeemed to receive priority review under an expedited timeframe for a subsequent marketing application for a different product. A PRV may also be sold or transferred from the initial sponsor to another sponsor and may be further transferred any number of times before it is used. On December 27, 2020, the Rare Pediatric Disease Priority Review Voucher Program was extended. Under the current statutory sunset provisions, after September 30, 2024, FDA may only award a voucher for an approved rare pediatric disease product application if the sponsor has rare pediatric disease designation for the drug or biologic, and that designation was granted by September 30, 2024. After September 30, 2026, FDA may not award any rare pediatric disease priority review vouchers. It is possible the authority for FDA to award rare pediatric disease priority review vouchers will be further extended by Congress.
Post-Approval Requirements
Any products for which we receive FDA approval are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Products may be promoted only for the approved indications and in accordance with the provisions of the approved label. Further, manufacturers must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Manufacturers and other entities involved in the manufacturing and distribution of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the product. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards, and test each product batch or lot prior to its release. Manufacturers and other parties involved in the drug supply chain for prescription drugs and biologics must also comply with product tracking and tracing requirements and for notifying the FDA of counterfeit, diverted, stole and intentionally adulterated products or products that are otherwise unfit for distribution in the United States.
75
The FDA may withdraw a product approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, untitled or warning letters, holds on clinical trials, product seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions or civil or criminal penalties.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, FDA regulations, guidance, and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration dates, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA; however, there can be no assurance that any such extension will be granted to us.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric exclusivity is another type of exclusivity in the United States. Pediatric exclusivity, if granted, provides an additional six months to an existing exclusivity or statutory delay in approval resulting from a patent certification. This six-month exclusivity, which runs from the end of other exclusivity protection or patent delay, may be granted based on the voluntary completion of a pediatric clinical trial in accordance with an FDA-issued “Written Request” for such a clinical trial.
76
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition—a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug in the United States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease or condition with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the United States for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same disease or condition, except in limited circumstances, such as if the second applicant demonstrates the clinical superiority of its product to the product with orphan drug exclusivity through a demonstration of superior safety, superior efficacy, or a major contribution to patient care. Orphan drug exclusivity does not prevent FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
Pediatric Information
Under the Pediatric Research Equity Act of 2003, as amended, NDAs or supplements to NDAs must contain data adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDCA requires that a sponsor who is planning to submit a marketing application for a drug product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days of an end-of-Phase 2 meeting or as may be agreed between the sponsor and the FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of data or full or partial waivers. The FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials, and/or other clinical development programs.
Disclosure of Clinical Trial Information
Sponsors of certain clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, trial sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
77
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we may obtain regulatory approval. In the United States, sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of coverage and reimbursement from third-party payors. Third-party payors include government authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the reimbursement rate that the payor will pay for the drug product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Moreover, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Third-party payors are increasingly challenging the price and examining the medical need and cost-effectiveness of medical products and services, in addition to their safety and efficacy. In order to obtain coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical need and cost-effectiveness of any products, in addition to the costs required to obtain regulatory approvals. Our product candidates may not be considered medically necessary or cost-effective. If third-party payors do not consider a product to be cost-effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit.
The U.S. government and state legislatures have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, collectively, the Affordable Care Act (“ACA”), contains provisions that may reduce the profitability of drug products, including, for example, increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on cost containment measures in the United States has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
78
Other Healthcare Laws and Compliance Requirements
If we obtain regulatory approval of our products, we will be subject to various federal and state laws targeting fraud and abuse in the healthcare industry. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business in the U.S. and applicable privacy legislation outside of the U.S., such as the GDPR in the European Union and the United Kingdom. The laws that may affect our ability to operate include:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order, or recommendation of, an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity can be found guilty of violating the statute without actual knowledge of the statute or specific intent to violate it. The term remuneration has been interpreted broadly to include anything of value. Further, courts have found that if “one purpose” of remuneration is to induce referrals, the federal Anti-Kickback statute is violated. Violations are subject to significant civil and criminal fines and penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs. In addition, a claim submitted for payment to any federal healthcare program that includes items or services that were made as a result of a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act (“FCA”). The Anti-Kickback Statute has been interpreted to apply to arrangements between biopharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers, among others, on the other. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, and on November 20, 2020, the Office of Inspector General, or OIG, finalized further modifications to the federal Anti-Kickback Statute; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, including the FCA and civil monetary penalty laws which impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement or record to avoid, decrease or conceal an obligation to pay money to the federal government. A claim that includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim under the FCA. Manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. The FCA also permits a private individual acting as a “whistleblower” to bring qui tam actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery or settlement. When an entity is determined to have violated the FCA, the government may impose civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes federal criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also created federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements or using or making any false or fraudulent document in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity can be found guilty of violating HIPAA fraud provisions without actual knowledge of the statute or specific intent to violate it; |
| ● | the federal transparency laws, including the provision of the ACA referred to as the federal Physician Payment Sunshine Act, that requires applicable manufacturers of covered drugs to disclose payments and other transfers of value provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain non-physician providers (such as physician assistants and nurse practitioners), and teaching hospitals and physician ownership and investment interests; |
79
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information on certain covered healthcare providers, health plans, and healthcare clearinghouses, known as covered entities, as well as their respective business associates; |
| ● | Federal government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner to government programs; |
| ● | Federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; and |
| ● | the General Data Protection Regulation (EU) 2016/679 in the European Union, which has been also been incorporated into United Kingdom’s laws, which includes among other obligations and requirements on: (i) when and how an organization can collect and rely upon consent obtained from an individual to process their personal data; (ii) the provision of information to individuals relating to the processing of their personal data; (iii) how to notify personal data breaches to competent data protection authorities and/or individuals; and (iv) may include additional requirements in relation to the processing of certain categories of data (i.e. “sensitive information” or special category data), which includes health data, data revealing racial or ethnic origin, and genetic data. |
Healthcare reform
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could impact our ability to sell our products profitably.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, (the “Medicare Modernization Act”) established the Medicare Part D program and provided authority for limiting the number of drugs that will be covered in any therapeutic class thereunder. The Medicare Modernization Act, including its cost reduction initiatives, could decrease the coverage and reimbursement rate that we or a collaborator receive for any of our approved products. Furthermore, private payors often follow Medicare coverage policies and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from the Medicare Modernization Act may result in a similar reduction in payments from private payors.
In March 2010, the Affordable Care Act (“ACA”) was enacted, which substantially changed the way health care is financed by both governmental and private insurers, and significantly impacted the U.S. biopharmaceutical industry. The ACA imposes a significant annual fee on companies that manufacture or import branded prescription drug products. It also contains substantial new provisions intended to broaden access to health insurance, reduce or constrain the growth of health care spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, and impose additional health policy reforms, any of which could negatively impact our business. Among other things, it subjects biological products to potential competition by lower-cost biosimilars, expands the types of entities eligible for the 340B drug discount program, introduced a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of certain branded prescription drugs, and created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D.
80
The ACA is likely to continue the downward pressure on pharmaceutical and medical device pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
Since its enactment, there have been judicial, Congressional and executive challenges to certain aspects of the ACA, including efforts to repeal or replace the ACA. For example, on June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA.
In addition, other legislative and regulatory changes have been proposed and adopted in the United States since the ACA was enacted. For example:
| ● | The U.S. Budget Control Act of 2011, among other things, included aggregate reductions of Medicare payments to providers of 2% per fiscal year. The reductions remain in effect through 2031. |
| ● | The U.S. American Taxpayer Relief Act of 2012 among other things, further reduced Medicare payments to several types of providers. |
| ● | The American Rescue Plan Act of 2021 eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024. |
| ● | Due to the Statutory Pay-As-You-Go Act of 2010, estimated budget deficit increases resulting from the American Rescue Plan Act of 2021, and subsequent legislation, Medicare payments to providers will be further reduced starting in 2025 absent further legislation. |
These laws and regulations may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices we may obtain for any of our product candidates for which we may obtain regulatory approval or the frequency with which any such product candidate is prescribed or used.
Additionally, there has been increasing legislative and enforcement interest in the United States with respect to drug pricing practices. There has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, and review the relationship between pricing and manufacturer patient programs. Further, the government has issued regulations and guidance for states to build and submit importation plans for drugs from outside the United States, pursuant to section 804 of the MMA. CMS has stated that drugs imported by states under this rule will not be eligible for federal rebates under Section 1927 of the Social Security Act and manufacturers would not report these drugs for “best price” or Average Manufacturer Price purposes. Since these drugs are not considered covered outpatient drugs, CMS further stated it will not publish a National Average Drug Acquisition Cost for these drugs. If further implemented, importation of drugs from Canada could materially and adversely affect the price we receive for any of our product candidates.
Additionally, on November 30, 2020, HHS published a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. Pursuant to court order, the removal and addition of the aforementioned safe harbors were delayed, and the inflation Reduction Act of 2022 delayed implementation of the rule until January 1, 2032. Although a number of these and other proposed measures may require authorization through additional legislation to become effective, and the Biden administration may reverse or otherwise change these measures, both the Biden administration and Congress have indicated that they will continue to seek new legislative and/or administrative measures to control drug costs.
81
In addition, the Inflation Reduction Act of 2022, or IRA includes several provisions that will impact our business to varying degrees, including provisions that reduce the out-of-pocket cap for Medicare Part D beneficiaries to $ 2,000 starting in 2025; impose new manufacturer financial liability on certain drugs in Medicare Part D, allow the U.S. government to negotiate Medicare Part B and Part D price caps for certain high-cost drugs and biologics without generic or biosimilar competition, require companies to pay rebates to Medicare for certain drug prices that increase faster than inflation, and delay the rebate rule that would limit the fees that pharmacy benefit managers can charge. The implementation of the IRA is currently subject to ongoing litigation challenging the constitutionality of the Medicare drug price negotiation program. The effects of the IRA on our business and the healthcare industry in general are not yet known.
We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we, or our collaborators, would receive for any approved drug, which could have an adverse effect on customers for our product candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors.
Regulation in the European Union
Product development, the regulatory approval process, and safety monitoring of medicinal products and their manufacturers in the European Union (“EU”) proceed in much the same manner as they do in the United States. Therefore, many of the issues discussed above apply similarly in the context of the EU. In addition, medicinal products are subject to the extensive price and reimbursement regulations of the various EU Member States.
Clinical Trials Approval
As is the case in the United States, the various phases of preclinical and clinical research in the EU are subject to significant regulatory controls.
In April 2014, the EU adopted the Clinical Trials Regulation (EU) No 536/2014 (Regulation), which replaced the previous Clinical Trials Directive 2001/20/EC (Directive) on January 31, 2022. The transitory provisions of the Regulation provide that by January 31, 2025, all ongoing clinical trials must have transitioned to the Regulation.
The Regulation overhauls the former system of approvals for clinical trials in the EU. Specifically, the Regulation, which is directly applicable in all Member States (meaning that no national implementing legislation in each Member State is required), aims at simplifying and streamlining the approval of clinical trials in the EU. For instance, the Regulation provides for a streamlined application procedure using a single entry point (the Clinical Trials Information System) and strictly defined deadlines for the assessment of clinical trial applications.
Following the end of the Brexit transition period, the Medicines and Healthcare products Regulatory Agency (“MHRA”), the UK medicines regulator, continues to authorize clinical trials in the UK. The UK has implemented the now-repealed Clinical Trials Directive into national law through the Medicines for Human Use (Clinical Trials) Regulations 2004 (as amended). The extent to which the regulation of clinical trials in the UK will mirror the EU Regulation in the long term is not yet certain, however in March 2023, the MHRA published a detailed response to the consultation on a set of proposals designed to improve and strengthen the UK clinical trials legislation, which ran in early 2022. The MHRA has stated they will now take forward new legislation to update the UK clinical trials legislation in line with the detailed response to the consultation. The key purpose of the new legislation will be to provide a more flexible regime to make it easier to conduct trials in the UK, increase the transparency of clinical trials conducted in the UK and make trials more patient centered.
82
PRIME Designation
PRIME is a voluntary scheme launched by the EMA to enhance support for the development of medicines that target an unmet medical need. The scheme focuses on medicines that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options. These medicines are considered priority medicines by the EMA. The scheme is open to medicines under development and for which the applicant intends to apply for a marketing authorization through the centralized procedure. This scheme is based on enhanced interaction and early dialogue with developers of promising medicines, to optimize development plans and speed up evaluation and enable accelerated assessment of medicines applications. Medicines under the PRIME scheme can expect to be eligible for accelerated assessment at the time of application for a marketing authorization. Where, during the course of development, a medicine no longer meets the eligibility criteria, support under the PRIME scheme may be withdrawn.
Acceptance into the PRIME scheme is based on the applicant’s ability to demonstrate that the medicine has potential to benefit patients with unmet medical needs based on early clinical data. Applicants from the academic sector and micro-, small- and medium-sized enterprises (“SMEs”) can apply earlier on the basis of compelling non-clinical data and tolerability data from initial clinical trials.
Once a candidate medicine has been selected for PRIME, the EMA will:
| ● | appoint a rapporteur from the Committee for Medicinal Products for Human Use (“CHMP”) or from the Committee for Advanced Therapies (“CAT”) for an advanced therapy, to provide continuous support and help to build knowledge ahead of a marketing authorization application (“MAA”); |
| ● | organize a kick-off meeting with the CHMP/CAT rapporteur and a multidisciplinary group of experts, so that they provide guidance on the overall development plan and regulatory strategy; |
| ● | assign a dedicated contact point; |
| ● | provide scientific advice at key development milestones, involving additional stakeholders such as health-technology-assessment bodies, to facilitate quicker access for patients to the new medicine; and |
| ● | confirm potential for accelerated assessment at the time of an application for marketing authorization. |
Marketing Approval
Marketing approvals under the EU regulatory system may be obtained through a centralized or national procedure. The centralized procedure results in the grant of a single marketing authorization that is valid for all—currently 27—European Union Member States and in the additional Member States of the European Economic Area, or EEA (Iceland, Liechtenstein and Norway).
The United Kingdom ceased to be a Member State of the EU on January 31, 2020. Further information on the marketing authorization regime in the United Kingdom is included in the Marketing Approval in the United Kingdom – Post-Brexit Regime section below.
Pursuant to Regulation (EC) No. 726/2004, as amended, the centralized procedure is mandatory for certain types of products, including products designated as orphan medicinal products pursuant to Regulation (EC) No. 141/2000, medicines produced by biotechnological processes, advanced-therapy medicinal products (gene-therapy, somatic cell-therapy or tissue-engineered medicines), and medicinal products containing a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto- immune and other immune dysfunctions and viral diseases. The CHMP also has the discretion to permit other products to use the centralized procedure if they contain a new active substance, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU.
In the MAA, the applicant has to properly and sufficiently demonstrate the quality, safety and efficacy of the product. Under the centralized procedure, the CHMP, possibly in conjunction with other committees, is responsible for drawing up the opinion of the EMA on any matter concerning the admissibility of the files submitted in accordance with the centralized procedure, such as an opinion on the granting, variation, suspension or revocation of a marketing authorization, and pharmacovigilance.
83
The CHMP and other committees are also responsible for providing guidance and have published numerous guidelines that may apply to our product candidates. These guidelines provide additional guidance on the factors that the EMA will consider in relation to the development and evaluation of products and may include, among other things, the preclinical studies required in specific cases; and the manufacturing and control information that should be submitted in an MAA; and post-approval measures required to monitor patients and evaluate the long term efficacy and potential adverse reactions. Although these guidelines are not legally binding, we believe that our compliance with them is likely necessary to gain approval for any of our product candidates.
The maximum timeframe for the evaluation of an MAA by the CHMP under the centralized procedure is 210 days after receipt of a valid application, excluding clock stops, where additional information or written or oral explanation is to be provided by the applicant in response to the questions asked by the CHMP. Clock stops may extend the timeframe of evaluation of an MAA considerably beyond 210 days. When an application is submitted for a marketing authorization in respect of a product which is expected to be of major public interest, particularly from the viewpoint of therapeutic innovation, the applicant may request an accelerated assessment procedure. If the CHMP accepts such request, the timeframe of 210 days for assessment will be reduced to 150 days (excluding clock stops) but it is possible that the CHMP can revert to the standard time-limit for the centralized procedure if it considers that the application is no longer appropriate to conduct an accelerated assessment.
If the CHMP concludes that the quality, safety and efficacy of the product is sufficiently proven, it adopts a positive opinion. This is sent to the European Commission which drafts a decision. After consulting with the Member States, the European Commission adopts a decision and grants a marketing authorization, which is valid for the whole of the EU. The marketing authorization may be subject to certain conditions, which may include, without limitation, the performance of post-authorization safety and/or efficacy studies.
EU legislation also provides for a system of regulatory data and market exclusivity. According to Article 14(11) of Regulation (EC) No. 726/2004, as amended, and Article 10(1) of Directive 2001/83/EC, as amended, upon receiving marketing authorization, innovative medicinal products approved on the basis of a complete and independent data package benefit from eight years of data exclusivity and an additional two years of market exclusivity. Data exclusivity prevents applicants for authorization of generics or biosimilars of these innovative products from referencing the innovator’s preclinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the EU, during a period of eight years from the date on which the reference product was first authorized in the EU. During the additional two-year period of market exclusivity, a generic or biosimilar MAA can be submitted and authorized, and the innovator’s data may be referenced, but no generic or biosimilar medicinal product can be placed on the EU market until the expiration of the market exclusivity. The overall ten-year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder, or MAH, obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. There is no guarantee that a product will be considered by the EMA to be an innovative medicinal product, and products may not qualify for data exclusivity. Even if a compound is considered to be an innovative medicinal product, so that the innovator is able to gain the period of data exclusivity, another company nevertheless could also market another version of the product if such company obtained a marketing authorization based on an MAA with a complete and independent data package of pharmaceutical tests, preclinical tests and clinical trials.
Additional rules apply to medicinal products for pediatric use under Regulation (EC) No. 1901/2006. Potential incentives include a six-month extension of any supplementary protection certificate granted pursuant to Regulation (EC) No. 469/2009, however not in cases in which the relevant product is designated as an orphan medicinal product pursuant to Regulation (EC) No. 141/2000, as amended. Instead, medicinal products designated as orphan medicinal products that qualify for additional protections under Regulation (EC) No. 1901/2006 may receive an extension of the ten-year market exclusivity period granted under Regulation (EC) No. 141/2000 to twelve years subject to the conditions applicable to orphan medicinal products.
84
Marketing Approval in the United Kingdom – Post-Brexit Regime
Now that the UK has left the EU, the MHRA is the UK’s stand-alone medicines and medical devices regulator, taking any decisions and carrying out any functions which were previously carried out at the EU-level by the EMA (except for decisions on MAAs made through the EU procedures to market products in Northern Ireland).
Great Britain is no longer covered by the EU’s procedures for the grant of marketing authorizations (under the Northern Ireland Protocol, Northern Ireland is covered by the centralized authorization procedure and can be covered as a Concerned Member State under the decentralized or mutual recognition procedures for the time being) and a separate marketing authorization is required to market products in Great Britain.
On January 1, 2024, a new international recognition framework was put in place, which may have regard to decisions on the approval of marketing authorizations made by the EMA and certain other regulators when considering an application for a Great Britain marketing authorization. The MHRA also has the power to have regard to marketing authorizations approved in European Union Member States through decentralized or mutual recognition (but not purely national procedures) procedures, with a view to granting marketing authorizations in the UK or Great Britain.
On February 27, 2023, the UK government and the European Commission announced a political agreement in principle to replace the Northern Ireland Protocol with a new set of arrangements, known as the “Windsor Framework”. This new framework fundamentally changes the existing system under the Northern Ireland Protocol, including with respect to the regulation of medicinal products in the UK. In particular, the MHRA will be responsible for approving all medicinal products destined for the UK market (Great Britain and Northern Ireland) and the EMA will no longer have any role in approving medicinal products destined for Northern Ireland. A single UK-wide marketing authorization will be granted by the MHRA for all medicinal products to be sold in the UK, enabling products to be sold in a single pack and under a single authorization throughout the UK. The Windsor Framework was approved by the EU-UK Joint Committee on March 24, 2023, so the UK government and the EU will enact legislative measures to bring it into law. On June 9, 2023, the MHRA announced that the medicines aspects of the Windsor Framework will apply from January 1, 2025.
Orphan Designation Regulation
In the EU, Regulation (EC) No. 141/2000, as amended, states that a product will be designated as an orphan medicinal product if its sponsor can establish:
| ● | that it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; |
| ● | either (i) such condition affects no more than five in ten thousand persons in the EU when the application is made, or (ii) that it is unlikely that the product, without the benefits derived from orphan status, would generate sufficient return in the EU to justify the necessary investment in its development; and |
| ● | that there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the EU or, if such method exists, the product would be of a significant benefit to those affected by that condition compared to products available for the condition. |
Regulation (EC) No. 847/2000 sets out further provisions for implementation of the criteria for designation of a product as an orphan medicinal product. An application for the designation of a products as an orphan medicinal product must be submitted at any stage of development of the product before filing of a MAA.
85
Orphan designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following the grant of a marketing authorization. During this market exclusivity period, neither the EMA nor the European Commission nor any of the competent authorities in the EU Members States can accept an application or grant a marketing authorization for a “similar medicinal product”. A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. This ten year period may however be reduced to six years if, at the end of the fifth year, it is established, with respect to the product concerned, that the criteria for orphan designation are no longer met, for example when it is shown on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity. Notwithstanding the foregoing, a marketing authorization may be granted, for the same therapeutic indication, to a similar medicinal product if:
| ● | the holder of the marketing authorization for the original orphan medicinal product has given its consent to the second applicant; |
| ● | the holder of the marketing authorization for the original orphan medicinal product is unable to supply sufficient quantities of the product; or |
| ● | the second applicant can establish in the application that the second product, although similar to the orphan medicinal product already authorized, is safer, more effective or otherwise clinically superior. |
Regulation (EC) No. 847/2000 lays down definitions of the concept of ‘clinical superiority’. Orphan designation does not shorten the duration of the regulatory review and approval process.
Since January 1, 2021, a separate process for orphan designation has applied in Great Britain. Currently, the requirements for orphan designation in Great Britain largely mirror those in the EU (save that they apply to the Great Britain market only – e.g. if the prevalence of the condition is no more than five in ten thousand persons in Great Britain). The main difference to the EU system is that there is no pre-marketing authorization orphan designation in Great Britain. Instead, the MHRA will make a decision on orphan status at the time it decides whether to approve the MAA.
As in the EU, medicinal products with orphan status will benefit from up to ten years of market exclusivity from the date of first approval of the product in Great Britain.
Manufacturing and Manufacturers’ License
Pursuant to Directive 2003/94/EC, as transposed into the national laws of the Member States, the manufacturing of investigational medicinal products and approved medicinal products is subject to a separate manufacturer’s license and must be conducted in strict compliance with cGMP requirements, which mandate the methods, facilities, and controls used in manufacturing, processing, and packing of products to assure their safety and identity. Manufacturers must have at least one qualified person permanently and continuously at their disposal in the EU. The qualified person is ultimately responsible for certifying that each batch of finished product released onto the market has been manufactured in accordance with cGMP and the specifications set out in the marketing authorization or investigational medicinal product dossier. cGMP requirements are enforced through mandatory registration of facilities and inspections of those facilities. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and subject the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil and criminal penalties.
The provisions of Directive 2003/94/EC, as transposed in the UK, have generally been preserved in the UK following the end of the Brexit Transition Period (subject to applicable amendments to ensure its effective operation in the post-Brexit context). The UK-EU Trade and Cooperation Agreement also contains provisions relating to the mutual recognition of cGMP inspections and documentation.
86
Advertising
In the EU and the UK, the promotion of prescription medicines is subject to intense regulation and control, including EU and national legislation as well as self-regulatory codes (“industry codes”). Advertising legislation inter alia includes a prohibition on direct-to-consumer advertising of prescription only medicines. All prescription medicines advertising must be consistent with the product’s approved summary of product characteristics, and must be factual, accurate, balanced and not misleading. Advertising of prescription medicines pre-approval or off-label is not allowed. Some jurisdictions require that all promotional materials for prescription medicines be subjected to either prior internal or external regulatory review and approval.
Other Regulatory Requirements
A holder of a marketing authorization for a medicinal product is legally obliged to fulfill a number of obligations by virtue of its status as an MAH. The MAH can delegate the performance of related tasks to third parties, such as distributors or marketing partners, provided that this delegation is appropriately documented and the MAH maintains legal responsibility and liability.
The obligations of an MAH include:
| ● | Manufacturing and batch release. MAHs should guarantee that all manufacturing operations comply with relevant laws and regulations, applicable good manufacturing practices, with the product specifications and manufacturing conditions set out in the marketing authorization and that each batch of product is subject to appropriate release formalities. |
| ● | Pharmacovigilance. MAHs are obliged to establish and maintain a pharmacovigilance system, including a qualified person responsible for oversight, submit safety reports to the regulators and comply with the good pharmacovigilance practice guidelines adopted by the EMA. |
| ● | Advertising and promotion. MAHs remain responsible for all advertising and promotion of its products, including promotional activities by other companies or individuals on their behalf and in some cases must conduct internal or regulatory pre-approval of promotional materials. Regulation in this area also covers interactions with healthcare practitioners and/or patient groups, and in some jurisdictions obligations to disclose such interactions exist. |
| ● | Medical affairs/scientific service. MAHs are required to disseminate scientific and medical information on its medicinal products to healthcare professionals, regulators and patients. |
| ● | Legal representation and distributor issues. MAHs are responsible for regulatory actions or inactions of their distributors and agents. |
| ● | Preparation, filing and maintenance of the application and subsequent marketing authorization. MAHs must maintain appropriate records, comply with the marketing authorization’s terms and conditions, fulfill reporting obligations to regulators, submit renewal applications and pay all appropriate fees to the authorities. |
Likewise, in the UK, a holder of a marketing authorization is subject to a number of ongoing obligations.
We may hold any future marketing authorizations granted for our product candidates in our own name, or appoint an affiliate or a collaboration partner to hold marketing authorizations on our behalf. Any failure by an MAH to comply with these obligations may result in regulatory action against an MAH and ultimately threaten our ability to commercialize our products.
87
Reimbursement
In the EU, the pricing and reimbursement mechanisms by private and public health insurers vary largely by country and even within countries. The public systems reimbursement for standard products is determined by guidelines established by the legislator or responsible national authority. The approach taken varies by EU Member State. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. Other EU Member States allow companies to fix their own prices for medicines, but monitor and control company profits and may limit or restrict reimbursement. The downward pressure on healthcare costs in general, particularly prescription products, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products and some EU countries require the completion of studies that compare the cost-effectiveness of a particular product candidate to currently available therapies in order to obtain reimbursement or pricing approval. Special pricing and reimbursement rules may apply to orphan medicinal products. Inclusion of orphan medicinal products in reimbursement systems tend to focus on the medical usefulness, need, quality and economic benefits to patients and the healthcare system as for any medicine. Acceptance of any medicinal product for reimbursement may come with cost, use and often volume restrictions, which again can vary by country. In addition, results based rules of reimbursement may apply.
The UK also has its own pricing and reimbursement rules.
The aforementioned EU rules are generally applicable in the EEA.
Reform of the Regulatory Framework in the European Union
The European Commission introduced legislative proposals in April 2023 that, if implemented, will replace the current regulatory framework in the EU for all medicines (including those for rare diseases and for children). The European Commission has provided the legislative proposals to the European Parliament and the European Council for their review and approval. In October 2023, the European Parliament published draft reports proposing amendments to the legislative proposals, which will be debated by the European Parliament. Once the European Commission’s legislative proposals are approved (with or without amendment), they will be adopted into EU law.
88
C. Organizational structure
At December 31, 2023, ProQR Therapeutics N.V. is the ultimate parent company of the following entities:
| ● | ProQR Therapeutics Holding B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics I B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics II B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics III B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics IV B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics V B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics VI B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics VII B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics VIII B.V. (the Netherlands, 100%); |
| ● | ProQR Therapeutics IX B.V. (the Netherlands, 100%); and |
| ● | ProQR Therapeutics I Inc. (United States, 100%). |
ProQR Therapeutics N.V. is also the statutory director of Stichting Bewaarneming Aandelen ProQR (“ESOP Foundation”) and has full control over this entity.
D. Property, Plants and Equipment
The Company leases office and laboratory facilities of 4,818 square meters at Zernikedreef in Leiden, the Netherlands, where our headquarters and our laboratories are located. The current lease agreement for these facilities terminates on June 30, 2031 and may be renewed for subsequent 5-year terms. The Company also leases office space in the United States, at CIC Cambridge. The office space is located at 245 Main Street, Cambridge, MA 02142. We believe that our existing facilities are adequate to meet current needs and that suitable alternative spaces will be available in the future on commercially reasonable terms.
Item 4A: Unresolved Staff Comments
Not applicable.
Item 5: Operating and Financial Review and Prospects
You should read the following discussion and analysis of our financial condition and results of operations together with our audited financial statements, including the notes thereto, included elsewhere in this Annual Report. The following discussion is based on our financial statements prepared in accordance with IFRS as issued by the IASB which might differ in material respects from generally accepted accounting principles in the United States. In addition to historical financial information, the following discussion and analysis includes forward-looking statements that involve risks, uncertainties and assumptions. See “Cautionary Note Regarding Forward-Looking Statements.” Our actual results and timing may differ materially from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those described under Item 3.D: “Risk factors” and elsewhere in this Annual Report.
89
A. Operating Results
Overview
To date, we have financed our operations primarily through our initial public and follow-on offerings, our ATM facility and private placements of equity securities, convertible loans, licensing and research collaborations and to a lesser extent through funding from patient organizations and governmental bodies, such as Rijksdienst voor Ondernemend Nederland (RVO – Netherlands Enterprise Agency).
In December 2022, the Company issued 9,381,586 shares to Eli Lilly and Company (“Lilly”) pursuant to the amended and restated licensing and research collaboration between the Company and Lilly, resulting in gross proceeds of € 14,122,000, with no significant transaction costs. In February 2023, ProQR also received an upfront payment of € 56,412,000.
In November 2021, the Company filed a shelf registration statement, which permitted: (a) the offering, issuance and sale by the Company of up to a maximum aggregate offering price of $ 300,000,000 of its ordinary shares, warrants and/or units; and (b) as part of the $ 300,000,000, the offering, issuance and sale by us of up to a maximum aggregate offering price of $ 75,000,000 of its ordinary shares that may be issued and sold under a sales agreement with Cantor Fitzgerald & Co in one or more at-the-market offerings. In 2023, 2022 and 2021, no shares were issued pursuant to this ATM facility.
In September 2021, the Company issued 3,989,976 shares to Lilly pursuant to the original licensing and research collaboration, resulting in gross proceeds of € 25,270,000, with no significant transaction costs. In addition, an up-front payment of € 16,849,000 was received in October 2021.
In April 2021, the Company consummated an underwritten public offering of 15,923,077 ordinary shares at an issue price of $ 6.50 per share. The gross proceeds from this offering amounted to € 88,115,000 while the transaction costs amounted to € 5,499,000, resulting in net proceeds of € 82,616,000.
At December 31, 2023, we had cash and cash equivalents of € 118,925,000. In 2023, our revenues consist of non-refundable upfront fees in connection with collaboration and license agreements. To date, we have not generated any revenues from royalties or product sales. Based on our current plans, we do not expect to generate royalty or product revenues for the foreseeable future.
We have generated losses since our inception in February 2012. For the years ended December 31, 2023, 2022 and 2021, we incurred net losses of € 27,735,000, € 64,204,000, and € 60,799,000, respectively. At December 31, 2023, we had an accumulated deficit of € 400,850,000. We expect to continue incurring losses for the foreseeable future as we invest in our Axiomer and Trident platforms and continue our preclinical studies of our product candidates.
Recent Accounting Pronouncements
There are no IFRS standards as issued by the IASB or interpretations issued by the IFRS interpretations committee that are effective for the first time for financial years beginning on or after January 1, 2023 that had a material impact on our financial statements.
A number of new standards, amendments to standards and interpretations are effective for annual periods beginning on after January 1, 2023 and have not been applied in preparing these consolidated financial statements. None of these are expected to have a material impact on the Company in the current or future reporting periods and on foreseeable future transactions. The Company does not plan to adopt these standards early.
90
Foreign Private Issuer Exemptions
As a foreign private issuer, we are not subject to the same requirements that are imposed upon U.S. domestic issuers by the Securities and Exchange Commission (“SEC”). Under the Exchange Act, we will be subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. For example, although we intend to report our financial results on a quarterly basis, we will not be required to issue quarterly reports, proxy statements that comply with the requirements applicable to U.S. domestic reporting companies, or individual executive compensation information that is as detailed as that required of U.S. domestic reporting companies. We will also have four months after the end of each fiscal year to file our annual reports with the SEC and will not be required to file current reports as frequently or promptly as U.S. domestic reporting companies. We may also present financial statements pursuant to IFRS instead of pursuant to U.S. GAAP. Furthermore, although the members of our management and supervisory boards will be required to notify the Dutch Authority for the Financial Markets of certain transactions they may undertake, including with respect to our ordinary shares, our officers, directors and principal shareholders will be exempt from the requirements to report transactions in our equity securities and from the short-swing profit liability provisions contained in Section 16 of the Exchange Act. These exemptions and leniencies will reduce the frequency and scope of information and protections available to you in comparison to those applicable to a U.S. domestic reporting companies.
Financial Operations Overview
Revenue
Revenues to date have consisted principally of non-refundable upfront fees and research and development service fees in connection with collaboration and license agreements.
Other Income
Other income mainly consists of the net proceeds from the Company’s divestment of its late-stage ophthalmic intellectual property assets, sepofarsen and ultevursen, to Laboratoires Théa (“Théa”), and grant income from government-related organizations and charities. (Government) Grants are recognized in other income in the same period in which the related research and development expenses are recognized.
Research and Development Expenses
Research and development expenses consist principally of:
| ● | salaries for research and development staff and related expenses, including social security costs and share-based compensation expenses; |
| ● | costs related to our preclinical and (former) clinical activities and trials, including costs paid to CROs; |
| ● | costs for production of (pre-)clinical compounds and drug substances by contract manufacturers, including supporting studies such as stability studies and other analysis of the compounds; |
| ● | costs of related facilities, materials and equipment; and |
| ● | depreciation of tangible fixed assets used to develop our product candidates. |
91
Our research and development expenses in 2023, 2022 and 2021 primarily related to the following key programs:
| ● | Axiomer |
Research and development expenses relating to our Axiomer platform, including expenses relating to the work performed under our research and collaboration agreement with Eli Lilly and Company (“Lilly”), primarily consist of salaries, costs for production of the preclinical compounds and costs paid to CROs for our preclinical studies. Other significant costs are our internal laboratory costs, including laboratory consumables and allocated housing expenses, as well as consultancy costs in support of our research and development activities for Axiomer.
| ● | Sepofarsen for the treatment of LCA |
In 2022 and 2021, the research and development costs relating to sepofarsen primarily consisted of salaries and costs paid to CROs for clinical studies, including statistical analyses, and manufacturing of process performance qualification drug substance batches as well as (pre-)commercial drug product batches. Other significant costs are our internal laboratory costs, including the materials used in the labs and rent allocated to the lab space, as well as consultancy costs in the support of our research and development activities. In 2023, costs related to sepofarsen were limited.
| ● | Ultevursen for the treatment of Usher syndrome |
In 2022 and 2021, the research and development costs relating to ultevursen primarily consisted of salaries, costs paid to CROs for managing the clinical study, costs for statistical analyses and manufacturing of process performance qualification drug substance batches. Other significant costs are our internal laboratory costs, including the materials used in the labs and rent allocated to the lab space, as well as consultancy costs in the support of our research and development activities. In 2023, costs related to ultevursen were limited.
For the years ended December 31, 2023, 2022 and 2021, we incurred research and development expenses of € 25,148,000, € 50,867,000 and € 42,220,000, respectively.
Our research and development expenses may vary substantially from period to period based on the timing of our research and development activities. Research and development expenses are expected to increase as we continue our joint research projects with Lilly and our investments in the Axiomer and Trident platforms, while progressing our internal pipeline targets towards clinical development.
Changes in internal or external variables with respect to the development of our Lilly collaboration program and the development of our Axiomer platform and resulting product candidates could result in a significant change in the costs and/or timing associated with the development of such product candidates. For example, if the FDA, EMA or other regulatory authority were to require us to conduct preclinical and clinical studies beyond those which we currently anticipate, or if we experience significant delays in enrollment in any future clinical trials, we could be required to expend significant additional financial resources and time on the completion of our development programs.
General and Administrative Expenses
Our general and administrative expenses consist principally of:
| ● | salaries for employees other than research and development staff and related expenses, including social security costs and share-based compensation expenses; |
| ● | professional fees for auditors and other consulting expenses not related to research and development activities; |
| ● | professional fees for lawyers; |
| ● | cost of facilities, communication and office expenses; |
92
| ● | IT expenses; and |
| ● | depreciation of property, plant and equipment not related to research and development activities. |
Since our IPO in September 2014 we have incurred additional costs associated with operating as a public company. These public company-related expenses include costs of additional personnel, additional legal fees, accounting and audit fees, managing directors’ and supervisory directors’ liability insurance premiums and costs related to investor relations. We expect that our general and administrative expenses will remain fairly stable in upcoming years.
Share-Based Compensation
Share-based compensation reflects the costs of our equity-settled share option plan, which are measured at the fair value of the options and restricted stock units at the grant date, and which are recognized over the course of each of the separate vesting tranches of the applicable vesting period of the options involved. The share-based compensation is recognized in the income statement, with a corresponding entry to the equity-settled employee benefits reserve, which is part of equity. See Note 13(d) to the financial statements included elsewhere in this Annual Report for additional information on share-based compensation.
Financial Income and Expense
To date, our cash and cash equivalents have been deposited primarily in savings and short-term deposit accounts. Savings and deposit accounts generate interest income (or a limited amount of interest expenses in 2022 and 2021). In 2023, 2022 and 2021 we held deposits in U.S. dollars.
Financial expenses primarily consist of interest expenses on convertible loans and government loans. Financial income and expense also includes foreign exchange gains or losses on our U.S. dollar denominated cash and cash equivalents and other foreign currency denominated monetary items.
Results related to derecognition of financial liabilities
Results related to derecognition of financial liabilities represent gains or losses arising from the extinguishment of convertible loans in 2023 and 2022.
Results related to financial liabilities measured at fair value through profit or loss
Results related to financial liabilities measured at fair value through profit or loss represent changes in the fair value of derivative financial instruments since their initial recognition. In 2023, 2022 and 2021, these derivative financial instruments consist of conversion options and warrants issued in connection with our convertible loans. Following the extinguishment of such loans in the third quarter of 2022, the related conversion options have been derecognized. The warrants issued to the convertible loan issuers have not yet expired at December 31, 2023 and therefore have not been derecognized.
Income Tax
Due to the operating losses incurred since inception, the Company has no income tax provisions as at December 31, 2023. Realization of deferred tax assets is dependent on future earnings, if any, the timing and amount of which are uncertain. Accordingly, we have not yet recognized any deferred tax asset related to operating losses.
93
Results of Operations
Comparison of the periods ended December 31, 2023 and 2022
The following table sets forth our results of operations for the periods indicated.
Year ended December 31, | ||||||
| 2023 |
| 2022 |
| Change | |
(€ in thousands) | ||||||
Revenue |
| 6,514 |
| 3,594 |
| 2,920 |
Other income |
| 3,011 |
| 765 |
| 2,246 |
Research and development costs |
| (25,148) |
| (50,867) |
| 25,719 |
General and administrative costs |
| (16,236) |
| (18,651) |
| 2,415 |
Operating result |
| (31,859) |
| (65,159) |
| 33,300 |
Financial income |
| 2,593 |
| 4,863 |
| (2,270) |
Financial expense | (1,458) | (5,127) | 3,669 | |||
Results related to associates | — | (8) | 8 | |||
Gain on disposal of subsidiary | 92 | — | 92 | |||
Results related to derecognition of financial liabilities | 1,866 | (1,390) | 3,256 | |||
Results related to financial liabilities measured at fair value through profit or loss | 953 | 2,713 | (1,760) | |||
Corporate income taxes | 78 | (96) | 174 | |||
Net loss |
| (27,735) |
| (64,204) |
| 36,469 |
Revenue
In 2023, we realized revenue from our license and research collaboration agreement with Lilly amounting to € 6,514,000 (2022: € 3,237,000). The increase in Lilly revenue is due to new projects under the Lilly collaboration that were started in 2023.
In 2023, we realized no revenue from our license and research collaboration agreement with Yarrow (2022: € 357,000). The decrease in Yarrow revenue is due to the termination of the Yarrow collaboration in the second quarter of 2022.
Other income
In 2023, other income consisted primarily of the net proceeds from the Company’s divestment of its late-stage ophthalmic intellectual property assets, sepofarsen and ultevursen, to Théa. No such income was recognized in 2022.
In 2022, other income included grant income from the Foundation Fighting Blindness (“FFB”) for the purpose of developing ultevursen. FFB grant income amounted to € 594,000 in 2022 while no such income was recognized in 2023.
Research and development costs
Research and development costs amounted to € 25,148,000 for the year ended December 31, 2023 compared to € 50,867,000 for the year ended December 31, 2022. These costs were primarily related to the development of our Axiomer platform, including costs incurred under the Lilly collaboration in 2023 and 2022. In 2022, the Company also incurred costs related to the sepofarsen and ultevursen clinical trials and the wind-down of those ophthalmology programs.
Our research and development expenses are highly dependent on the development phases of our product candidates. Research and development expenses are expected to increase as we continue our joint research projects with Lilly and our investments in the Axiomer and Trident platforms, while progressing our internal pipeline targets towards clinical development.
94
The decrease in research and development costs in the year ended December 31, 2023 compared to the year ended December 31, 2022 includes the effects of:
| ● | Lower costs of CROs for the Phase 2/3 clinical trials for ultevursen. The trials were wound down in the second half of 2022 and wind-down costs were recognized in 2022. The Company incurred very limited further wind-down costs in 2023; |
| ● | Lower employee benefits (excluding share-based compensation) in 2023 compared to 2022, resulting from the effects of a reorganization in 2022, partly offset by a company-wide inflationary correction on salaries; |
| ● | Advisory costs relating to the Phase 2/3 clinical trials for ultevursen that we incurred in 2022 but not in 2023; |
| ● | The above effects are partly offset by increased expenses related to the development of our Axiomer platform in 2023. |
General and administrative costs
General and administrative costs amount to € 16,236,000 for the year ended December 31, 2023 and € 18,651,000 for the year ended December 31, 2022. The decrease in general and administrative costs in the year ended December 31, 2023 compared to the year ended December 31, 2022 includes the effects of:
| ● | Lower employee benefits (excluding share-based compensation) resulting from the effects of a reorganization in 2022, partly offset by a company-wide inflationary correction on salaries; |
| ● | The above effect is partly offset by increased share-based compensation, reflecting the higher value of grants of share options to general and administrative staff. |
Financial income
We had financial income of € 2,593,000 for the year ended December 31, 2023, as compared to € 4,863,000 for the year ended December 31, 2022. The financial income mainly reflects interest income in 2023 and foreign exchange gains on cash and cash equivalents denominated in U.S. dollars in 2022.
Financial expenses
Financial expenses amounted to € 1,458,000 for the year ended December 31, 2023, as compared to € 5,127,000 for the year ended December 31, 2022. The decrease in 2023 compared to 2022 is mainly due to lower interest expenses in 2023 following the extinguishment of our convertible debt financing agreement with Kreos and Pontifax in September 2022.
Results related to derecognition of financial liabilities
In 2023, results related to derecognition of financial liabilities consist of a gain of € 1,866,000 (2022: € 1,390,000) resulting from waiver agreements that ProQR’s subsidiary Amylon Therapeutics B.V. (“Amylon”) entered into with all of its lenders. Such lenders’ loan agreements with Amylon are severed and any claims to repayment of any outstanding debt and accumulated interest are renounced.
In 2022, results related to derecognition of financial liabilities also included a loss of € 2,534,000 on the extinguishment of the Pontifax and Kreos loans, containing accelerated interest payments and early repayment penalties.
95
Results related to financial liabilities measured at fair value through profit or loss
Results related to financial liabilities measured at fair value through profit or loss amounted to a gain of € 953,000 for the year ended December 31, 2023, as compared to a gain of € 2,713,000 for the year ended December 31, 2022. These results relate to fair value changes in our derivative financial instruments, consisting of issued warrants. The gains in 2023 and 2022 were mainly due to the decrease in ProQR’s share price.
Comparison of the periods ended December 31, 2022 and 2021
Reference is made to our 2022 annual report on form 20-F, filed on March 29, 2023, for a comparison of the periods ended December 31, 2022 and 2021.
B. Liquidity and Capital Resources
To date, we have financed our operations primarily through our initial public and follow-on offerings, our ATM facility and private placements of equity securities, convertible loans, licensing and research collaborations and to a lesser extent through funding from patient organizations and governmental bodies, such as Rijksdienst voor Ondernemend Nederland (RVO – Netherlands Enterprise Agency).
In December 2022, the Company issued 9,381,586 shares to Lilly pursuant to the amended and restated licensing and research collaboration between the Company and Lilly, resulting in gross proceeds of € 14,122,000, with no significant transaction costs. In February 2023, ProQR also received an upfront payment of € 56,412,000.
In November 2021, the Company filed a shelf registration statement, which permitted: (a) the offering, issuance and sale by the Company of up to a maximum aggregate offering price of $ 300,000,000 of its ordinary shares, warrants and/or units; and (b) as part of the $ 300,000,000, the offering, issuance and sale by us of up to a maximum aggregate offering price of $ 75,000,000 of its ordinary shares that may be issued and sold under a sales agreement with Cantor Fitzgerald & Co in one or more at-the-market offerings. In 2023, 2022 and 2021, no shares were issued pursuant to this ATM facility.
In September 2021, the Company issued 3,989,976 shares to Lilly pursuant to the original licensing and research collaboration, resulting in gross proceeds of € 25,270,000, with no significant transaction costs. In addition, an up-front payment of € 16,849,000 was received in October 2021.
In April 2021, the Company consummated an underwritten public offering of 15,923,077 ordinary shares at an issue price of $ 6.50 per share. The gross proceeds from this offering amounted to € 88,115,000 while the transaction costs amounted to € 5,499,000, resulting in net proceeds of € 82,616,000.
As at December 31, 2023 the Company’s outstanding shares totaled 81,354,592. The following items may result in future dilution for our shareholders:
| ● | 1,054,010 treasury shares held by the Company, which can be used for all general purposes including option exercises under the employee stock option plan; |
| ● | 1,839,782 treasury shares held by the ESOP foundation, which can be used for option exercises under the employee stock option plan; |
| ● | 12,203,189 shares authorized for issuance for future grants under the employee stock option plan; |
| ● | 679,628 warrants held by former convertible debt holders. |
If all of the above items were converted into shares at December 31, 2023, the number of outstanding shares would be 97,131,201.
96
Cash Flows
The table below summarizes our statement of cash flows for the years ended December 31, 2023 and 2022.
Year ended December 31, | ||||||
| 2023 |
| 2022 |
| Change | |
(€ in thousands) | ||||||
Net cash generated by / (used in) operating activities |
| 21,548 |
| (68,508) |
| 90,056 |
Net cash generated by / (used in) investing activities |
| 4,278 |
| (702) |
| 4,980 |
Net cash used in financing activities |
| (2,275) |
| (30,890) |
| 28,615 |
Net increase / (decrease) in cash and cash equivalents |
| 23,551 |
| (100,100) |
| 123,651 |
Currency effect cash and cash equivalents |
| 599 |
| 7,351 |
| (6,752) |
Cash and cash equivalents at the beginning of the period |
| 94,775 |
| 187,524 |
| (92,749) |
Cash and cash equivalents at the end of the period |
| 118,925 |
| 94,775 |
| 24,150 |
Net cash generated by operating activities amounted to € 21,548,000 in the year ended December 31, 2023, whereas net cash used in operating activities amounted to € 68,508,000 in the year ended December 31, 2022. The Company experienced a net positive cash flow from operating activities in 2023 mainly because of the receipt of the Lilly up-front payment of € 56,412,000 in February 2023. In addition, total operating costs decreased by € 28,134,000 in 2023 compared to 2022, which had a further positive impact on net cash generated by operating activities.
Net cash generated by investing activities in the year ended December 31, 2023 is mainly driven by the divestment of our late-stage ophthalmic intellectual property assets, sepofarsen and ultevursen, to Théa. Under the terms of the agreement, ProQR received an initial payment of € 8,000,000 and incurred cash outflows directly related to the transaction amounting to € 2,351,000 in 2023, leading to a net cash inflow from the Théa transaction of € 5,649,000 in 2023. This is partly offset by net investments in laboratory equipment and other fixed assets amounting to € 1,311,000, leading to net cash generated by investing activities of € 4,278,000 in 2023. In the year ended December 31, 2022, net cash used in investing activities of € 702,000 primarily consisted of investments in laboratory equipment.
Net cash used in financing activities amounted to € 2,275,000 in the year ended December 31, 2023, compared to net cash used in financing activities amounting to € 30,890,000 in the year ended December 31, 2022. In 2023, net cash used in financing activities included repayments of the lease liability for the Company’s Leiden headquarters amounting to € 1,621,000 and the partial repayment amounting to € 1,008,000 of the Innovation Credit loan from RVO. In September 2022, ProQR extinguished its debt with Pontifax and Kreos by repaying all outstanding principal amounts, resulting in a cash outflow of € 43,372,000. In December 2022, the Company issued 9,381,586 shares to Lilly pursuant to the amended and restated licensing and research collaboration between the Company and Lilly, resulting in gross proceeds of € 14,122,000, with no significant transaction costs. In 2022, net cash used in financing activities also included repayments of the lease liability of € 1,674,000.
Reference is made to our 2022 annual report on form 20-F, filed on March 29, 2023, for a comparison of the periods ended December 31, 2022 and 2021.
Cash and Funding Sources
The table below summarizes our main sources of financing for the years ended December 31, 2023, 2022 and 2021.
| Equity |
| Convertible |
| Government | Lilly Upfront |
| |||
| Capital | Loans | Borrowing | Payments | Total | |||||
(€ in thousands) | ||||||||||
Year ended December 31, 2021 |
| 108,477 | 26,520 | 1,137 | 16,849 |
| 152,983 | |||
Year ended December 31, 2022 |
| 14,122 | — | — | — | 14,122 | ||||
Year ended December 31, 2023 |
| — | — | — | 56,412 | 56,412 | ||||
Total |
| 122,599 |
| 26,520 |
| 1,137 | 73,261 |
| 223,517 | |
97
Equity capital
In 2023, we did not raise any equity capital.
In 2022, our main source of funding consisted of the 9,381,586 shares issued to Lilly resulting in proceeds of € 14,122,000. The shares were issued at a discount of € 450,000 compared to the prevailing share price at the transaction date. The gross amount of € 14,572,000 excluding the discount was recognized in equity.
In April 2021, we executed an underwritten public offering of 15,923,077 ordinary shares, resulting in net proceeds of € 82,616,000. In addition, in September 2021, we issued 3,989,976 shares to Lilly pursuant to the original licensing and research collaboration, resulting in net proceeds of € 25,270,000.
Convertible loans and government borrowing
In 2023 and 2022, we did not raise any funding from convertible loans and government borrowing.
In December 2021, the Company drew down a loan of $ 26,520,000 under its loan agreement with Pontifax and Kreos. In September 2022, ProQR extinguished its debt with Pontifax and Kreos by repaying all outstanding principal amounts.
In 2021, we also received funding from the Dutch government, through its agency Rijksdienst voor Ondernemend Nederland (RVO – Netherlands Enterprise Agency), under the Innovation credit program for sepofarsen, amounting to € 1,137,000.
At December 31, 2023, we had borrowings (including accrued interest) of € 4,292,000, which wholly consisted of borrowings from a government body. At December 31, 2022, we had borrowings (including accrued interest) of € 6,771,000, which consisted of borrowings from a government body (€ 4,942,000) and convertible loans (€ 1,829,000). Cash is denominated in both U.S. dollars and euros.
Lilly upfront payments
In 2023 and 2021, our sources of funding included the receipt of upfront payments under the collaboration agreement with Lilly, which are reflected in changes in working capital. In 2022, we did not receive upfront payments from Lilly. The upfront payment received in 2023 amounted to € 56,412,000 and the upfront payment received in 2021 amounted to € 16,849,000.
For a description of our financial commitments, see below.
Funding Requirements
Our material cash requirements from known contractual and other obligations include contractual commitments to repay borrowings, lease liabilities, and trade and other payables. In 2024, such cash requirements include the following undiscounted amounts:
| ● | Lease liabilities amounting to € 2,288,000 |
| ● | Trade and other payables amounting to € 11,709,000 |
Beyond 2024, our cash requirements include the following undiscounted amounts:
| ● | Borrowings amounting to € 4,583,000 |
| ● | Lease liabilities amounting to € 16,223,000 |
We expect that our cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements into mid-2026. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. Our present and future funding requirements will depend on many factors, including, among other things:
| ● | the investments required to further develop our Axiomer platform and the results of current and potential future collaborations involving the Axiomer technology. |
98
| ● | the progress, timing, resumption and completion of preclinical testing and clinical trials for our current or any future product candidates; |
| ● | the number of potential new product candidates we identify and decide to develop; |
| ● | the costs involved in growing our organization to the size needed to allow for the research, development and potential commercialization of our current or any future product candidates; |
| ● | the costs involved in filing patent applications and maintaining and enforcing patents or defending against claims or infringements raised by third parties; |
| ● | the time and costs involved in obtaining regulatory approval for our product candidates and any delays we may encounter as a result of evolving regulatory requirements or adverse results with respect to any of these product candidates; |
| ● | any licensing or milestone fees we might have to pay during future development of our current or any future product candidates; |
| ● | the amount of revenues, if any, we may derive from upfront, milestone or royalty payments resulting from licensing and research collaboration agreements. |
For more information as to the risks associated with our future funding needs, see Item 3.D: “Risk Factors”.
Capital Expenditures
The following table sets forth our capital expenditures for the years ended December 31, 2023, 2022 and 2021:
Year ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(€ in thousands) | ||||||
Purchases of tangible fixed assets |
| 1,371 |
| 708 |
| 718 |
Purchases of tangible fixed assets primarily consist of investments in laboratory equipment. Such investments increased in 2023 compared to 2022 and 2021 as the Company increased its in-house oligonucleotide manufacturing capacity.
Commitments
Rent
Since 2012, the Company is domiciled in Leiden, the Netherlands. We are currently a party to a lease agreement for laboratory and office space in Leiden. The commitment for lease liabilities that is disclosed under ‘Funding requirements' above consists of our commitment under this laboratory and office space lease agreement.
Patent license agreements
On October 29, 2018, ProQR signed an agreement with Ionis Pharmaceuticals (“Ionis”) to license QR-1123 (formerly “IONIS-RHO-2.5Rx”), an RNA medicine for autosomal dominant retinitis pigmentosa (“adRP”) caused by the P23H mutation in the rhodopsin (“RHO”) gene. Under the terms of the agreement, ProQR was granted an exclusive worldwide license to QR-1123 and relevant patents. In 2018, ProQR paid the first installment of an upfront payment in ordinary shares in the aggregate amount of $ 2,500,000 at $ 22.23 per share, which represents a 20% premium (based on the volume weighted average price of the previous 20 trading days) to its common stock, to Ionis upon signing the agreement. In 2019, ProQR paid the second installment of the upfront payment in ordinary shares in the aggregate amount of $ 3,501,000, at $ 9.43 per share. This license agreement was terminated effective January 2024.
99
In April 2014, the Company entered into a Patent License Agreement with Radboud University Medical Center (“Radboud”) in the field of antisense oligonucleotide-based therapy for Leber congenital amaurosis (“LCA’). Under the terms of this license agreement, the Company has an exclusive, sublicensable, world-wide royalty-bearing license under certain Radboud patent rights to develop, make, have made, use, sell, offer for sale and import certain licensed products of Radboud for use in all prophylactic and therapeutic uses in the field of LCA. This license is assigned in full per December 2023 as part of the divestment of the product sepofarsen.
In June 2015, the Company entered into another license agreement with Radboud. Under the terms of this license agreement, the Company has an exclusive, sublicensable, world-wide royalty-bearing license under certain Radboud patent rights to develop, make, have made, use, sell, offer for sale and import certain licensed products of Radboud for use in all prophylactic and therapeutic uses in the field of Usher syndrome. This license was assigned in full per December 2023 as part of the divestment of the product ultevursen.
In January 2018, the Company entered into a license agreement with Inserm Transfert SA and Assistance-Publique-Hôpiteaux de Paris. Under the terms of the agreement, the Company has a world-wide, exclusive, royalty-bearing license under patent rights belonging to Inserm Transfert SA and other co-owners to develop, have developed, make, have made, use, have used and sell, have sold or otherwise distribute certain licensed products related to antisense oligonucleotides for treating LCA and method of treatment claims relating to modulation of the splicing of the CEP290 gene product. This license agreement is assigned per December 2023 in connection with the sale of sepofarsen. In consideration for the assignment, the Company has agreed to accept certain royalty obligations upon sepofarsen reaching certain regulatory milestones and net sales of products sold.
In January 2017, the Company entered into an agreement with the Leiden University Medical Center (“LUMC”), which gives us a world-wide, exclusive, royalty-bearing license in the field of Huntington’s disease, under certain patent rights of LUMC regarding antisense oligonucleotide based therapies. This license agreement contains certain diligence obligations for the Company coupled to milestone payments and complements the Company’s intellectual property relating to the HD program. This license is terminated per July 2023.
In February 2019, the Company entered into an agreement with the University of Rochester, New York, which gives us a world-wide, exclusive, royalty-bearing, sublicensable license in the field of antisense oligonucleotides for use in nucleotide specific RNA editing through pseudouridylation, under certain patent rights of University of Rochester. This license agreement contains certain diligence obligations for the Company coupled to milestone payments and complements the Company’s intellectual property relating to the Axiomer/pseudouridylation program.
In September 2020, the Company entered into an agreement with Vico Therapeutics B.V., which gives us a world-wide, exclusive, royalty-bearing, sublicensable license in the field of the prophylactic and therapeutic use of antisense oligonucleotide for the treatment of Fuch’s Endothelial Corneal Dystrophy caused by a trinucleotide repeat, under certain patent rights of Vico Therapeutics B.V. In partial consideration of the rights and licenses granted by the license agreement, the Company is required to make annual maintenance payments. Unless terminated earlier in accordance with this license agreement, the agreement will stay in effect until the expiration of all of the licensed patent rights. The license agreement may be terminated by either party in the event of an uncured breach by the breaching party. Vico Therapeutics B.V. may terminate the license agreement if the Company applies for an order or an order is made declaring the Company bankrupt or granting the Company suspension of payments, or a liquidator is appointed for the Company, or the Company is dissolved, liquidated, or ceases to carry on all or a substantial part of its business or a decision is taken to that effect, or in the event uncured payment defaults. The development of this candidate has been suspended per the strategic shift in focus as announced in August 2022.
Refer to Item 4.B: “Business Overview” for more details on patent license agreements.
Clinical support agreements
On February 9, 2018, the Company entered into an agreement with FFB, under which FFB has provided funding of $ 6.8 million (€ 6.3 million) to advance ultevursen into the clinic.
100
Pursuant to the terms of the agreement, we were obligated to make certain repayments to FFB subject to development milestones. In December 2023, upon the occurrence of the sale of ultevursen to Théa, these payables were settled by means of a lump-sum payment in the amount of € 1.1 million and a percentage of earn-out payments for milestones and sales to be received by us from Théa, ranging from 5-10%.
Research and development commitments
The Company has committed itself to a number of obligations amounting to € 8,893,000 at December 31, 2023 (2022: € 8,030,000). Of these obligations an amount of € 7,162,000 is due in 2024 and the remainder is due in 2 to 5 years.
Our commitments are set out in more detail in notes 25 and 26 to the financial statements as included elsewhere in this Annual Report.
C. Research and Development
See Item 4.B: “Business Overview” and Item 5: “Operating and Financial Review and Prospects”.
D. Trend Information
Other than as disclosed elsewhere in this Annual Report, we are not aware of any trends, uncertainties, demands, commitments or events for the period from January 1, 2023 to December 31, 2023 that are reasonably likely to have a material adverse effect on the Company’s net income, profitability, liquidity or capital resources, or that caused the reported financial information to be not necessarily indicative of future operating results or financial conditions. For a discussion of trends, see Item 5.A: “Operating results”.
E. Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which we have prepared in accordance with IFRS as issued by the IASB. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. Actual results may differ from these estimates under different assumptions or conditions. There have been no material adjustments to prior period estimates for any of the periods included in this Annual Report. Significant estimates and judgements are disclosed in disclosure Note 2 Basis of preparation to the financial statements, under (e) Use of estimates and judgements.
Our significant accounting policies are fully described in the notes to our financial statements appearing elsewhere in this Annual Report.
Item 6: Directors, Senior Management and Employees
A. Directors and senior management
We have a two-tier board structure consisting of our management board (‘raad van bestuur’) and a separate supervisory board (‘raad van commissarissen’). Below is a summary of relevant information concerning our supervisory board, management board and senior management.
Supervisory Board
The following table sets forth information with respect to each of our supervisory board members and their respective dates of birth. The terms of office of all our supervisory board members expire according to a rotation schedule drawn up by our supervisory board. The business address of our supervisory board members is our registered office address at Zernikedreef 9, 2333 CK Leiden, the Netherlands.
101
Our supervisory board is currently composed of the following members, all of whom are independent under applicable Nasdaq standards and all of whom are independent under the Dutch Corporate Governance Code (“DCGC”), with the exception of Ms. Theresa Heggie. Ms. Heggie was, prior to her appointment on our supervisory board in 2023, employed by ProQR as Chief Commercial Officer and Chief Operations Officer. Having been employed by the company within three years prior to her appointment on our supervisory board, Ms. Heggie does not qualify as independent under applicable Nasdaq standards and under the DCGC. We do not intend to follow Nasdaq’s requirements regarding Nasdaq Listing Rule 5605(b)(2), which mandates that independent directors must meet at regularly scheduled executive sessions where only independent directors are present.
Name |
| Date of Birth |
| Position |
| Member Since | Term expires | ||||||||||
Dinko Valerio | August 3, 1956 | Member of the Supervisory Board (Chair) | January 1, 2014 | 2024 | |||||||||||||
Alison F. Lawton | September 26, 1961 | Member of the Supervisory Board | September 17, 2014 | 2026 | |||||||||||||
James Shannon | June 5, 1956 | Member of the Supervisory Board | June 21, 2016 | 2024 | |||||||||||||
Bart Filius | July 5, 1970 | Member of the Supervisory Board | May 21, 2019 | 2027 | |||||||||||||
Begoña Carreño | December 13, 1971 | Member of the Supervisory Board | May 18, 2023 | 2027 | |||||||||||||
Theresa Heggie | November 17, 1960 | Member of the Supervisory Board | May 18, 2023 | 2027 | |||||||||||||
The following sets forth biographical information regarding our supervisory board members. There are no family relationships among the members of our supervisory board, management board or executive officers.
Dinko Valerio is one of our founders and currently serves as the chairman of our supervisory board which he joined in 2014. As a scientist and an experienced biotech entrepreneur Mr. Valerio is founder and former CEO of Crucell N.V., and one of the founders of its spinout, Galapagos Genomics. He was founder and former general partner of Aescap Venture, a life sciences venture capital firm and co-founder and current board member of Leyden Laboratories. In 1992 Mr. Valerio was appointed professor of gene therapy at the University of Leiden, where he also received his Ph.D. with honors.
Alison F. Lawton has served on our supervisory board since 2014. Ms. Lawton is an executive leader with more than 35 years of experience in biopharma. Most recently, she served as President and CEO of Kaleido Biosciences, Inc. Ms. Lawton previously served as Chief Operating Officer of Aura Biosciences, OvaScience and X4 Pharmaceuticals. She worked at various positions of increasing responsibility at Genzyme, and subsequently at Sanofi-Aventis, including as head of Genzyme Biosurgery and Global Market Access. Ms. Lawton currently serves on the board of directors of public pharmaceutical companies X4 Pharmaceuticals, and Dianthus Therapeutics, and the private companies AgBiome, SwanBio and BlueRock Therapeutics. She previously served on the boards of Verastem, CoLucid until its acquisition by Eli Lilly and Cubist Pharmaceuticals until its acquisition by Merck & Co. She is past President and Chair of the Board of Regulatory Affairs Professional Society and past FDA Advisory Committee member for Cell and Gene Therapy Committee. She earned her BSc in Pharmacology, with honors, from King’s College London.
James Shannon has served on our supervisory board since June 2016 and has been Chair of our Scientific Advisory Board since 2020. Mr. Shannon has had an extensive career in drug development and pharma. From 2012 until his retirement in 2015, he was Chief Medical Officer at GlaxoSmithKline. Prior to that he was Global Head of Pharma Development at Novartis and Senior Vice-President, Clinical Development at Sterling Winthrop Pharmaceuticals. He has previously held board positions at companies including Biotie, Circassia, Crucell, Endocyte and Cerimon Pharmaceuticals. Mr. Shannon currently is Chairman of the Board at Mannkind Corp and Kyowa Kirin NA and holds board positions at Horizon Pharma, myTomorrows and Leyden Labs. He received his undergraduate and postgraduate degrees at Queen’s University of Belfast and is a member of the Royal College of Physicians.
Bart Filius has served on our supervisory board since 2019. He joined Galapagos in 2014 as Chief Financial Officer and added the role of Chief Operating Officer in 2017. He was promoted to President and Chief Operating Officer in 2021. Prior to joining Galapagos, Mr. Filius held a variety of executive positions at Sanofi, where he was Vice President, Chief Financial Officer Europe, Country manager for The Netherlands and Vice President for Mergers & Acquisitions. Prior to joining Sanofi, Mr. Filius was a strategy consultant at Arthur D. Little. Mr. Filius has an MBA degree from INSEAD and a bachelor’s degree in business from Nyenrode University.
102
Begoña Carreño, PhD joined the ProQR Supervisory Board in 2023. Dr. Carreño is currently the Chief Business Development Officer at Vectura Fertin Pharma in Switzerland. Prior to this, she spent 18 years at Novartis Pharma AG in the Corporate BD&L group, her last role being World Wide BD&L Head in the Ophthalmology Franchise, based in Basel, Switzerland. Dr. Carreño has over 20 years Pharmaceutical Development experience. She is a seasoned & energetic BD&L professional that has led the BD&L efforts at Novartis across 5 different therapeutic franchises in the last 15 years. She has proven track record in licensing deals, M&A as well as developing collaborations within cross functional, multi-cultural, matrix environment at global, regional and country level. Before joining Novartis, she was the Head of External Pharmaceutical projects at Almirall (Barcelona, Spain). Dr. Carreño holds a PhD in Drug Delivery from the London School of Pharmacy (UK) and a BSc in Biochemistry from Keele University (UK).
Theresa Heggie was reappointed to our supervisory board in 2023. Previously, Ms. Heggie served as the Chief Operating Officer at ProQR, after originally joining the Management Team in 2021 as the Chief Commercial Officer. Prior to ProQR, she served as Chief Executive Officer of Freeline Therapeutics. She had senior commercial and operating roles at Alnylam Pharmaceuticals as Senior Vice President, Head of CEMEA and Shire where she built the EMEA rare disease business. Earlier in her career, Ms. Heggie held increasingly senior positions in the commercial organizations at Janssen Pharmaceuticals and Baxter Healthcare. She currently serves on the Board of BioCryst and previously served on our supervisory board from 2019-2021. She earned her BSc from Cornell University.
Management Board
The following table sets out information with respect to our management board members, their dates of birth and their positions at the company as of the date of this Annual Report. The business address of our management board members is our registered office address at Zernikedreef 9, 2333 CK Leiden, the Netherlands.
Name |
| Date of Birth |
| Position |
| Date of Appointment |
| Term Expires |
|
|---|---|---|---|---|---|---|---|---|---|
Daniel de Boer | April 12, 1983 | Chief Executive Officer | February 21, 2012 | 2026 | |||||
Rene Beukema | March 26, 1964 | Chief Corporate Development Officer and General Counsel | June 30, 2022 | 2026 |
The following sets forth biographical information regarding our management board members.
Daniel de Boer is our Founder and has served as our Chief Executive Officer since our incorporation in 2012. Mr. De Boer is a serial entrepreneur and passionate advocate for rare disease patients. After one of his children was diagnosed with a rare disease, he started ProQR to develop RNA therapies for rare diseases. Before founding ProQR, Mr. De Boer was founder and Chief Executive Officer of several technology companies. He is also strategic advisor at Hybridize Therapeutics, Meatable, Algramo, Xinvento, Avanzanite, BioColl Labs and a member of the advisory board at the Termeer Foundation. In 2018 Mr. De Boer was named "Emerging Entrepreneur of the Year" by EY. In 2019, Mr. De Boer was selected for the Young Global Leader program at the World Economic Forum.
René Beukema rejoined ProQR in 2022 having previously served as our Chief Corporate Development Officer and General Counsel from 2013 to 2018. Mr. Beukema is a seasoned M&A and equity capital markets executive and an experienced corporate lawyer. From 2019 until June 2022, Mr. Beukema held the Position of Chief Corporate Development Officer & General Counsel at Frame Therapeutics, a neoantigen immune-oncology biotechnology company. He was instrumental in financing Frame Therapeutics and selling it to CureVac, a Nasdaq Listed biotechnology company. From 2021 to 2024 Mr. Beukema was a Board Member of Fibriant BV, a biotechnology company focused on the development of technology and products based on recombinant human fibrinogen and thrombin. Prior to his initial tenure at the Company, he served as General Counsel and Corporate Secretary of Crucell for twelve years, following his positions as Senior Legal Counsel at GE Capital / TIP Europe and Legal Counsel at TNT Express Worldwide. Mr. Beukema was also a venture partner of Aescap Venture, a life sciences venture capital firm from 2011 to 2012 and is co-founder of myTomorrows, a Dutch life sciences company. He holds a post-doctoral degree in corporate law from the University of Nijmegen in co-operation with the Dutch Association of In-house Counsel (‘Nederlands Genootschap van Bedrijfsjuristen’) and a master's degree in Dutch law from the University of Amsterdam.
103
Officers
Our management board is supported by our officers. The following table sets forth information with respect to each of the officers, their respective dates of birth and their positions as of the date of this Annual Report. The business address of our officers is our registered office address at Zernikedreef 9, 2333 CK Leiden, the Netherlands.
Name |
| Date of Birth |
| Position |
|
|---|---|---|---|---|---|
Gerard Platenburg | February 24, 1964 | Chief Scientific Officer | |||
Sheila Sponselee | May 22, 1984 | Chief People and Operations Officer | |||
Jurriaan Dekkers | March 17, 1976 | Chief Financial Officer |
Gerard Platenburg is our co-founder and our Chief Scientific Officer. Mr. Platenburg has an extensive background in RNA modulation and orphan drug discovery and development and is currently in charge of our Innovation unit. Mr. Platenburg has more than twenty-five years of senior managerial experience in growing biotech companies. Prior to joining our company, Mr. Platenburg worked at Isa Pharmaceuticals B.V. as its Chief Executive Officer. Mr. Platenburg co-founded Prosensa Holding N.V., growing it to become a well-known clinical stage RNA modulation company, and held various positions including Chief Executive Officer and Chief Development Officer. Mr. Platenburg also worked at Pharming B.V. He is a passionate and driven pioneer of early-stage technologies. Mr. Platenburg has a master’s degree in chemistry and molecular biology from Leiden University in 1987 and pursued Ph.D. work at Leiden University. Mr. Platenburg is currently a scientific advisory board member at Hybridize Therapeutics.
Sheila Sponselee became Chief People and Operations Officer in 2023. In this role she leads Human Resources, Recruitment, Information Technology, Facilities and the Project Management Office. Ms. Sponselee joined ProQR in 2020 as VP of Human Resources and became VP, Head of People and Operations in 2022. Ms. Sponselee has more than 15 years of experience in human resources, recruitment, and operations across varied industries, including information technology and biopharma. Prior to joining ProQR, Ms. Sponselee most recently led Human Resources at MyTomorrows in the Netherlands from 2019 to 2020 and she managed Human Resources at Korton Group B.V. from 2017 to 2019. She studied human resource management at the University of Applied Science (BSc) and completed leadership programs at IMD Business School for Management and Leadership.
Jurriaan Dekkers joined ProQR in 2022 as Chief Financial Officer where he leads the finance and procurement functions. He brings to the role more than 20 years of experience in finance, management and leadership roles in biopharma and healthcare companies, as well as in audit and consulting services. Prior to joining ProQR he most recently served as CFO of AstraZeneca in the Netherlands and CEO of Acerta Pharma (part of the AstraZeneca Group) from 2018 to September 2022. Previously, Mr. Dekkers held various global and European finance roles at amongst others DaVita Medical Group, a US-listed international healthcare provider, from 2015 to 2018, and started his career at PwC. He is a Supervisory Board member at ‘Stichting Kinderpostzegels’ in the Netherlands. Mr. Dekkers holds a MSc. in Economics from the Erasmus University Rotterdam, the Netherlands (2001) and is a Certified Public Auditor (Register Accountant, “RA”) graduate from the Erasmus University Rotterdam (2004).
B. Compensation
Under Dutch law and our articles of association, the general meeting of shareholders must upon the proposal of our supervisory board adopt the compensation policy for the management board, which includes the outlines of the compensation of any members who serve on our management board. On June 30, 2022, the general meeting of shareholders adopted the current compensation policy of our company with respect to our management board as well as with respect to our supervisory board. The supervisory board determines the compensation of the management board members in accordance with the compensation policy. A proposal by the supervisory board with respect to compensation schemes in the form of shares or rights to shares is submitted for approval by the supervisory board to the general meeting of shareholders. Such proposal must set out at least the maximum number of shares or rights to shares to be granted to the management board and supervisory board, including the criteria for granting such shares or changes to such grants. The general meeting of shareholders may grant compensation to members of the supervisory board. The supervisory board will be reimbursed for their expenses.
104
Compensation of the Supervisory Board
The remuneration of the supervisory board members in 2023 is set out in the table below:
2023 | ||||||||
| Short term |
| Post-employment |
| Share-based |
| ||
employee benefits | benefits | payment | Total | |||||
(€ in thousands) | ||||||||
Mr. Dinko Valerio |
| 74 | — | 76 | 150 | |||
Mr. Antoine Papiernik* |
| — | — | — | — | |||
Ms. Alison F. Lawton |
| 50 | — | 76 | 126 | |||
Mr. James Shannon |
| 56 | — | 76 | 132 | |||
Mr. Bart Filius | 49 | — | 78 | 127 | ||||
Ms. Begoña Carreño** | 50 | — | 34 | 84 | ||||
Ms. Theresa Heggie*** | 30 | — | 241 | 271 | ||||
| 309 |
| — |
| 581 |
| 890 | |
Share-based payment reflects the fair value of share options granted under our Option Plan, as required by and determined in accordance with IFRS 2 - Share based payment (refer to Note 13(d) to the financial statements included elsewhere in this Annual Report).
* Mr. Papiernik stepped down from the supervisory board on May 18, 2023. In 2023, Mr. Papiernik waived his compensation.
** Ms. Carreño was elected to the supervisory board on May 18, 2023. The remuneration set forth for Ms. Carreño in the table above covers the period from May 18, 2023 to December 31, 2023.
*** Ms. Heggie was elected to the supervisory board on May 18, 2023. The remuneration set forth for Ms. Heggie in the table above covers the period from May 18, 2023 to December 31, 2023. Ms. Heggie’s share-based payments include the effects of options and RSUs that were granted to her before her reappointment to the supervisory board on May 18, 2023.
Under the current compensation principles, members of our supervisory board receive a board fee of € 34,000 per year and the chairperson receives a board fee of € 63,000 per year. In addition, the audit committee chairperson receives € 15,000 per year for service on that committee, and each other member of the audit committee receives € 7,000 per year for service on that committee. The compensation, nominating and corporate governance committee chairperson receives € 12,000 per year for service on that committee, and each other member of the compensation, nominating and corporate governance committee receives € 5,500 per year for service on that committee. The chairperson of the research and development committee receives € 12,000 per year for service on that committee, and each other member of the research and development committee receives € 5,500 per year for service on that committee. In addition, members of our supervisory board may be granted options with an underlying value of € 143,000 per year.
Compensation of the Management Board
The table below sets out a breakdown of the compensation in 2023 of the current members of the management board:
2023 | ||||||||
| Short term |
| Post-employment |
| Share-based |
| ||
| employee benefits |
| benefits | payment | Total | |||
| (€ in thousands) | |||||||
Mr. D.A. de Boer |
| 1,167 | 27 | 1,245 |
| 2,439 | ||
Mr. R.K. Beukema | 892 | 23 | 395 |
| 1,310 | |||
Share-based payment reflects the fair value of share options granted under our Option Plan during the year, as required by and determined in accordance with IFRS 2 - Share based payment (refer to Note 13(d) to the financial statements included elsewhere in this Annual Report).
105
Short-term employee benefits include an annual bonus of € 643,000 for Mr. de Boer and an annual bonus of € 481,000 for Mr. Beukema. In accordance with our compensation policy, the annual bonus was determined by the supervisory board and related to pre-determined quantified financial targets, non-financial/personal targets and/or company goals that were set by the supervisory board prior to the start of 2023. Each year the non-financial targets and goals of the company are derived from our strategic and organizational priorities and also include qualitative targets that are relevant for the responsibilities of Mr. de Boer and Mr. Beukema. Moreover, the size of the annual share-based payments of Mr. de Boer and Mr. Beukema is determined by the supervisory board based on contribution to the strategy of the Company, the Company’s long-term development, individual performance and alignment of total compensation to the median of the compensation reference group selected in accordance with our compensation policy.
For further detail on compensation of members of our supervision board, management board and senior management, see Note 27 to the financial statements included elsewhere in this Annual Report.
C. Board Practices
Supervisory Board
Our supervisory board is responsible for the supervision of the activities of our management board and our company’s general affairs and business. Our supervisory board may, also on its own initiative, provide the management board with advice and may request any information from the management board that it deems appropriate. In performing its duties, the supervisory board is required to act in the interests of our company (including its stakeholders) and its associated business as a whole. The members of the supervisory board are not authorized to represent us in dealings with third parties.
Pursuant to Dutch law, members of the supervisory board must be natural persons. Under our articles of association, the number of supervisory board members is determined by our supervisory board itself, provided there will be at least three supervisory board members. Our articles of association provide that members of the supervisory board are appointed by the general meeting of shareholders upon a binding nomination by the supervisory board. Our general meeting of shareholders may at all times deprive such a nomination of its binding character by a resolution passed by at least two-thirds of the votes cast representing more than 50% of our issued share capital, following which our supervisory board shall draw up a new binding nomination.
Our supervisory board rules provide that members of our supervisory board will serve for a maximum duration of three terms of four years. Our articles of association provide that the supervisory board members must retire periodically in accordance with a rotation schedule adopted by the supervisory board. A supervisory board member who retires in accordance with the rotation schedule can be reappointed immediately. The supervisory board appoints a chairman from among its members.
Under our articles of association, the general meeting of shareholders may suspend or remove supervisory board members at any time. A resolution of our general meeting of shareholders to suspend or remove a supervisory board member may be passed by a simple majority of the votes cast, provided that the resolution is based on a proposal by our supervisory board. In the absence of a proposal by our supervisory board, a resolution of our general meeting of shareholders to suspend or remove a supervisory board member shall require a majority of at least two-thirds of the votes cast representing more than 50% of our issued share capital.
In a meeting of the supervisory board, each supervisory board member is entitled to cast one vote. A supervisory board member may grant a written proxy to another supervisory board member to represent him at a meeting of the supervisory board. All resolutions by our supervisory board are adopted by a simple majority of the votes cast unless our supervisory board rules provide otherwise. In case of a tie in any vote of the supervisory board, the chairman of the supervisory board shall have the casting vote. Our supervisory board may also adopt resolutions outside a meeting, provided that such resolutions are adopted in writing, all supervisory board members are familiar with the resolution to be passed and provided that no supervisory board member objects to such decision-making process.
106
Management Board
Our management board is responsible for the day-to-day management of our operations under the supervision of the supervisory board. The management board is required to:
| ● | keep the supervisory board informed in a timely manner in order to allow the supervisory board to carry out its responsibilities; |
| ● | consult with the supervisory board on important matters; and |
| ● | submit certain important decisions to the supervisory board for its approval. |
Our management board may perform all acts necessary or useful for achieving our corporate purposes, other than those acts that are prohibited by law or by our articles of association. The management board as a whole and any management board member individually, are authorized to represent us in dealings with third parties. The management board may delegate this authority in whole or in part to employees of the company.
Under our articles of association, the number of management board members is determined by the supervisory board, and the management board must consist of at least one member. The supervisory board elects a CEO from among the members of the management board.
Members of the management board are appointed by the general meeting of shareholders upon a binding nomination of the supervisory board. Our general meeting of shareholders may at all times deprive such a nomination of its binding character by a resolution passed by at least two-thirds of the votes cast representing more than 50% of our issued share capital, following which our supervisory board shall draw up a new binding nomination.
Our management board rules provide that, unless the resolution appointing a management board member provides otherwise, members of our management board will serve for a maximum term of four years. Our articles of association provide that the management board members must retire periodically in accordance with a rotation schedule adopted by the management board. A management board member who retires in accordance with the rotation schedule may be reappointed immediately for a term of not more than four years at a time.
Board Diversity Matrix (as of the date of this Annual Report)
The table below summarizes certain self-identified information regarding the diversity of our supervisory directors as of the date of this Annual Report.
Country of Principal Executive Offices | Netherlands |
Foreign Private Issuer | Yes |
Disclosure Prohibited by Dutch Law | No |
Total Number of Directors | 6 |
Female | Male | Non-Binary | Did Not Disclose Gender | |
Part I: Gender Identity | ||||
Number of Directors | 3 | 3 | - | - |
Part II: Demographic Background | Number of Directors in Each Category | |||
Underrepresented Individual in Home Country Jurisdiction | - | |||
LGBTQ+ | - | |||
Did Not Disclose Demographic Background | 6 | |||
107
Service Agreements
We have entered into service agreements with our Chief Executive Officer, Daniel de Boer, and with our Chief Corporate Development Officer and General Counsel, René Beukema. The service agreement with Mr. de Boer contains a termination notice period of two months and the service agreement with Mr. Beukema contains a termination notice period of one month. The service agreements may be terminated in the event of an urgent reason (‘dringende reden’) at any time, without advance notice. The service agreements with Mr. de Boer and Mr. Beukema provide for a lump-sum payment following a change in control subject to certain conditions. Such payment is equal to 24 months of the individual’s monthly gross fixed salary in effect at the time of the change in control. The service agreements also contain certain restrictive covenants, including post termination non-competition and non-solicitation covenants, which survive for a period of 12 months post-termination.
Committees of the Supervisory Board
During 2023, our supervisory board had an audit committee, a compensation, nominating and corporate governance committee, and a research and development committee, each of which has an adopted charter.
Audit Committee
Our audit committee consists of Bart Filius (chair), Alison F. Lawton and Begoña Carreño. Each member satisfies the independence requirements of the Nasdaq listing standards and Rule 10A-3(b)(1) under the Exchange Act, and each member meets the criteria for independence set forth in best practice provision 2.1.8 of the DCGC. Bart Filius qualifies as an “audit committee financial expert,” as defined by the SEC in Item 16A: “Audit Committee Financial Expert” and as determined by our supervisory board. The audit committee oversees our accounting and financial reporting processes and the audits of our financial statements. The audit committee is responsible for, among other things:
| ● | making recommendations to our supervisory board regarding the appointment by the general meeting of shareholders of our independent auditor(s); |
| ● | overseeing the work of our independent auditor(s), including resolving disagreements between the management board, the officers and the independent auditors relating to financial reporting; |
| ● | pre-approving all audit and non-audit services permitted to be performed by the independent auditor(s); |
| ● | reviewing the independence and quality control procedures of the independent auditor(s); |
| ● | discussing material off-balance sheet transactions, arrangements and obligations with the management board and the independent auditor(s); |
| ● | reviewing and approving all proposed related-party transactions; |
| ● | discussing the annual audited statutory financial statements with the management board; |
| ● | annually reviewing and reassessing the adequacy of our audit committee charter; |
| ● | meeting separately with the independent auditors to discuss critical accounting policies, recommendations on internal controls, the auditor’s engagement letter and independence letter and other material written communications between the independent auditors and the management board and the officers; and |
| ● | attending to such other matters as are specifically delegated to our audit committee by our supervisory board |
108
| from time to time. |
Compensation, Nominating and Corporate Governance Committee
Our compensation, nominating and corporate governance committee consists of Theresa Heggie (chair), Dinko Valerio and James Shannon. Each member satisfies the independence requirements of the Nasdaq listing standards, and each member meets the criteria for independence set forth in best practice provision 2.1.8 of the DCGC, in each case, with the exception of Ms. Theresa Heggie. Ms. Heggie was, prior to her appointment to our supervisory board in 2023, employed by ProQR as Chief Commercial Officer and Chief Operations Officer from October 2021 to October 2022. Considering her employment by the company within three years prior to her appointment to our supervisory board, Ms. Heggie does not qualify as independent under applicable Nasdaq standards and under the DCGC. We do not intend to follow Nasdaq’s requirements regarding the independence of all members of the compensation, nominating and corporate governance committee. Dutch law and the DCGC do not require that the compensation, nominating and corporate governance committee be composed entirely of independent directors and only requires a mere majority. In accordance with Dutch law and the DCGC, our compensation, nominating and corporate governance committee consists of a majority of members who qualify as independent under applicable Nasdaq standards and under the DCGC. With respect to compensation matters, the compensation, nominating and corporate governance committee assists our supervisory board in reviewing and approving or recommending our compensation structure, including all forms of compensation relating to our supervisory board members, our management board members and our officers. The CEO may not be present at any compensation committee meeting while their compensation is deliberated. With respect to nominating and corporate governance matters, the compensation, nominating and corporate governance committee assists our supervisory board in selecting individuals qualified to become our supervisory board members and management board members, in determining the composition of the management board, supervisory board and its committees and our officers and in developing and recommending a set of corporate governance guidelines applicable to the Company. Subject to and in accordance with the terms of the compensation policy approved by our general meeting of shareholders from time to time, as required by Dutch law, the compensation, nominating and corporate governance committee is responsible for, among other things:
| ● | reviewing and making recommendations to the supervisory board with respect to compensation of our management board and supervisory board members; |
| ● | reviewing and approving the compensation, including equity compensation, change-of-control benefits and severance arrangements, of our officers (not part of our management board or supervisory board) as it deems appropriate; |
| ● | overseeing the evaluation of our management board members and our officers; |
| ● | reviewing periodically and making recommendations to our supervisory board with respect to any incentive compensation and equity plans, programs or similar arrangements; |
| ● | exercising the rights of our supervisory board under any equity plans, except for the right to amend any such plans unless otherwise expressly authorized to do so; |
| ● | attending to such other matters as are specifically delegated to our compensation committee by our supervisory board from time to time; |
| ● | approving the compensation package for the officers; |
| ● | periodically reviewing, in consultation with our CEO, our management board and our officers succession planning; |
| ● | recommending to the supervisory board persons to be nominated for election or re-election to the supervisory board and the management board at any meeting of the shareholders; |
109
| ● | overseeing the supervisory board’s annual review of its own performance and the performance of its committees; and |
| ● | considering, preparing, and recommending to the supervisory board a set of corporate governance guidelines. |
Our supervisory board may also delegate certain tasks and powers under our Option Plan to the compensation, nominating and corporate governance committee.
Research and Development Committee
Our research and development committee consists of James Shannon (chair), Dinko Valerio and Alison F. Lawton. Each member satisfies the independence requirements of the Nasdaq listing standards. In addition, each member meets the criteria for independence set forth in best practice provision 2.1.8 of the DCGC. The research and development committee assists the supervisory board in overseeing our product pipeline and research and development strategy. The research & development committee is responsible for, among other things:
| ● | reviewing the company’s research and development strategy, including the long-term strategy goals and objectives; |
| ● | reviewing and assessing quality of the research and development programs; |
| ● | reviewing the progress of the product pipeline, including a review and analysis of the progress and results of pre-clinical studies and clinical trials; |
| ● | reviewing and advising the management board about strategic opportunities to enhance innovation and development; |
| ● | reviewing and assessing scientific activities critical to the success of the company’s research and development strategy; and |
| ● | organizing and chairing meetings with the Company’s scientific advisory board for supporting its review and assessment the company’s research and development strategy. |
Insurance and Indemnification of Management Board and Supervisory Board Members
Under Dutch law, management board members, supervisory board members and certain other representatives may be held liable for damages in the event of improper or negligent performance of their duties. They may be held jointly and severally liable for damages to the company for infringement of the articles of association or of certain provisions of the Dutch Civil Code. They may also be liable towards third parties for infringement of certain provisions of the Dutch Civil Code. In certain circumstances they may also incur additional specific civil and criminal liabilities.
Our articles of association provide that we will indemnify our management board members, supervisory board members, former management board members and former supervisory board members (each an “Indemnified Person”) against (i) any financial losses or damages incurred by such Indemnified Person and (ii) any expense reasonably paid or incurred by such Indemnified Person in connection with any threatened, pending or completed suit, claim, action or legal proceedings, whether civil, criminal, administrative or investigative and whether formal or informal, in which he or she becomes involved, to the extent this relates to his or her position with the Company, in each case to the fullest extent permitted by applicable law. No indemnification shall be given to an Indemnified Person (a) if a Dutch court has established, without possibility for appeal, that the acts or omissions of such Indemnified Person that led to the financial losses, damages, suit, claim, action or legal proceedings result from either an improper performance of his duties as an officer of the Company or an unlawful or illegal act and (b) to the extent that his or her financial losses, damages and expenses are covered by an insurance and the insurer has settled these financial losses, damages and expenses (or has indicated that it would do so). Our supervisory board may stipulate additional terms, conditions and restrictions in relation to such indemnification.
110
D. Employees
As at December 31, 2023, we had a total of 156.6 employees (converted to FTE). Of these employees, 122.4 were engaged in research and development and 34.2 in general and administrative activities. For additional details we refer to Note 20 to the financial statements included elsewhere in this Annual Report. As far as we know, our employees are not represented by a labor union. We have never experienced any employment related work stoppages, and we consider our relations with our employees to be good. For more information as to the risks associated with our workforce reduction, see Item 3.D: “Risk Factors”.
E. Share Ownership
Refer to Item 7.A: “Major Shareholders” in this Annual Report.
F. Disclosure of a registrant’s action to recover erroneously awarded compensation
Not applicable.
Item 7: Major Shareholders and Related Party Transactions
| A. | Major Shareholders |
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as at December 31, 2023 by:
| ● | each of the members of our supervisory board and management board; and |
| ● | each person, or group of affiliated persons, known by us to beneficially own more than 5% of our ordinary shares. |
The percentage of shares beneficially owned is based on a total of 81,354,592 ordinary shares outstanding as at December 31, 2023. The numbers and percentages of ordinary shares beneficially owned are reported on the basis of regulations of the SEC governing the determination of beneficial ownership of securities. Under the applicable SEC rules, a person is deemed to be a “beneficial owner” of a security if that person has or shares voting power, which includes the power to vote or direct the voting of such security, or investment power, which includes the power to dispose of or to direct the disposition of such security, or to receive the economic benefit of ownership of the securities. A person is also deemed to be a beneficial owner of any securities of which that person has a right to acquire beneficial ownership within 60 days of December 31, 2023, and such securities are considered outstanding for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person. Under these rules, more than one person may be deemed beneficial owner of the same securities and a person may be deemed to be a beneficial owner of securities as to which such person has no economic interest. Except as otherwise indicated, each of the beneficial owners has, to our knowledge, sole voting and investment power with respect to the indicated ordinary shares, subject to community property laws, where applicable. Except as otherwise set forth below, the address of the beneficial owner is c/o ProQR Therapeutics N.V., Zernikedreef 9, 2333 CK, Leiden, the Netherlands.
111
Shares Beneficially Owned |
| ||||
Name and Address of Beneficial Owner |
| Number |
| Percentage |
|
Major Shareholders: |
|
|
|
| |
Eli Lilly & Co. 1 |
| 13,371,562 |
| 16.4 | % |
Van Herk 2 | 8,170,254 | 10.0 | % | ||
Privium / Stichting Aescap 3 |
| 5,406,159 |
| 6.6 | % |
Supervisory Board Members and Management Board Members |
|
|
| ||
Dinko Valerio 4 |
| 883,926 |
| 1.1 | % |
Theresa Heggie 5 |
| 255,505 |
| 0.3 | % |
James Shannon 6 |
| 228,852 |
| 0.3 | % |
Alison F. Lawton 7 |
| 171,054 |
| 0.2 | % |
Bart Filius 8 |
| 72,418 |
| 0.1 | % |
Begoña Carreño 9 | 6,841 | — | % | ||
Daniel de Boer 10 |
| 3,412,480 |
| 4.2 | % |
Rene Beukema 11 | 1,099,226 | 1.4 | % | ||
All supervisory board members and management board members as a group |
| 6,130,302 |
| 7.6 | % |
| 1 | Information is based on a report on Schedule 13G/A filed by Eli Lilly and Company (“Lilly”), an Indiana corporation, on December 23, 2022. 9,381,586 shares were issued to Lilly in December 2022, as part of the Company amended and restated research and collaboration agreement with Lilly, increasing the percentage of our shares held by Lilly from 5.6% to 16.4%. The address of Lilly is Lilly Corporate Center, Indianapolis, Indiana 46285. |
| 2 | Information is based on a report on Schedule 13G/A filed on February 14, 2024 by (i) Van Herk Investments B.V (“VHI”), with respect to Ordinary Shares beneficially owned by it, (ii) Van Herk Investments THI B.V. (“VHIT”), with respect to Ordinary Shares beneficially owned by VHI, (iii) Van Herk Private Equity Investments B.V. (“VHPI”), with respect to Ordinary Shares beneficially owned by VHI and VHIT, (iv) Stichting Administratiekantoor Penulata (“Penulata”), with respect to Ordinary Shares beneficially owned by VHI, VHIT and VHPI, (v) Van Herk Management Services B.V. (“VHMS”), with respect to Ordinary Shares beneficially owned by VHI, VHIT and VHPI, (vi) Onroerend Goed Beheer- en Beleggingsmaatschappij A. van Herk B.V. (“OGBBA”), with respect to Ordinary Shares beneficially owned by VHI, VHIT, VHPI and VHMS, (vii) A. van Herk Holding B.V. (Holdings), with respect to Ordinary Shares beneficially owned by VHI, VHIT, VHPI, VHMS and OGBBA, (viii) Stichting Administratiekantoor Abchrys (“Abchrys”), with respect to Ordinary Shares beneficially owned by VHI, VHIT, VHPI, VHMS, OGBBA and Holdings, and (ix) Adrianus van Herk (“Mr. van Herk”) with respect to Ordinary Shares beneficially owned by VHI, VHIT, VHPI, VHMS, OGBBA, Holdings, Penulata and Abchrys. Mr. van Herk is (i) an investor, (ii) the holder of all of the depositary receipts issued by Penulata and Abchrys, (iii) the sole board member of Penulata and Abchrys, and (iv) the sole managing director of VHMS, OGBBA and Holdings. Penulata holds substantially all of the issued and outstanding shares of VHPI. VHPI is the sole shareholder of VHIT. VHIT is the sole shareholder of VHI. Abchrys holds substantially all of the issued and outstanding shares of Holdings. Holdings is the sole shareholder of OGBBA. OGBBA is the sole shareholder of VHMS. VHMS is the sole managing director of VHI, VHIT and VHPI. The address of each of Mr. van Herk, VHI, VHIT, VHPI, Penulata, VHMS, OGBBA, Holdings and Abchrys is Lichtenauerlaan 30, 3062 ME Rotterdam, the Netherlands. |
| 3 | Information is based on a report on Schedule 13G filed on February 9, 2023, by Stichting Aescap Life Sciences (Aescap Life Sciences), Privium Fund Management B.V. (Privium), as the fund manager of Aescap, Stichting Aescap Genetics (Aescap Genetics), Inspirational Visions B.V. (Inspirational Visions) and Patrick Johan Hendrik Krol (Krol), the portfolio manager for Privium and Aescap Genetics and the managing director of Inspirational Visions. The total number of shares beneficially owned consists of 4,765,659 ordinary shares directly held by Aescap Life Sciences, 578,787 ordinary shares directly held by Aescap Genetics, 35,411 ordinary shares directly held by Inspirational Visions and 26,302 ordinary shares held by Patrick Johan Hendrik Krol. The address of each of Aescap Life Sciences, Aescap Genetics, Privium and Krol is Gustav Mahlerplein 3, 1082 MS Amsterdam, The Netherlands. The address of Inspirational Visions is A.J. Ernststraat 595-C, 1082 LN Amsterdam, The Netherlands. |
112
| 4 | Consists of 725,692 ordinary shares and options to acquire 158,234 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. Also includes 304,963 shares held by the Valerio family foundation, Stichting Administratiekantoor Endavit, for which Mr. Valerio is the managing director. The address of Stichting Administratiekantoor Endavit is Keizersgracht 290A, 1016 EW, Amsterdam, the Netherlands. |
| 5 | Consists of 26,499 ordinary shares and options to acquire 229,006 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. |
| 6 | Consists of 61,538 ordinary shares and options to acquire 167,314 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. |
| 7 | Consists of options to acquire 171,054 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. |
| 8 | Consists of options to acquire 72,418 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. |
| 9 | Consists of options to acquire 6,841 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023 |
| 10 | Consists of 705,309 ordinary shares and options to acquire 2,707,171 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. |
| 11 | Consists of 460,000 ordinary shares and options to acquire 639,226 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. |
| 12 | Consists of 1,979,038 ordinary shares and options to acquire 4,151,264 ordinary shares that are currently exercisable or will be exercisable within 60 days after December 31, 2023. |
Holdings by U.S. Shareholders
As at December 31, 2023, 99.98% of our ordinary shares were held by record holders in the United States, which excludes shareholders whose shares were held in nominee or street name by brokers.
B. Related Party Transactions
Since January 1, 2023, there have been no material transactions that are unusual in their nature or conditions with any of our related parties, except for transactions as set out in Note 27 to the financial statements included elsewhere in this Annual Report.
C. Interests of Experts and Counsel
Not applicable.
Item 8: Financial Information
A. Consolidated Statements and Other Financial Information
Consolidated financial statements
Consolidated financial statements are filed as part of this Annual Report, starting page F-1 and incorporated herein by reference.
113
Legal proceedings
From time to time we could be involved in legal proceedings that arise in the ordinary course of business. As at December 31, 2023, we believe no proceedings exists of which the outcome, if determined adversely, would have a material adverse effect on our financial position. During the period covered by the audited and approved financial statements contained herein, we have not been a party to or paid any damages in connection with litigation that has had a material adverse effect on our financial position. Any future litigation may result in substantial costs and be a distraction to management and employees. No assurance can be given that future litigation will not have a material adverse effect on our financial position. Refer to Item 3.D: “Risk factors.”
Dividends and other Distributions
We may only make distributions to our shareholders and other persons entitled to distributable profits, to the extent that our shareholders’ equity exceeds the sum of the paid-up and called-up share capital plus the reserves as required to be maintained by Dutch law or by our articles of association.
Under our articles of association, a (cumulative) dividend is first paid out of the profit, if available for distribution, on any preferred shares. Any amount remaining out of the profit is carried to reserve as the management board determines. After reservation by the management board of any profit, the remaining profit will be at the disposal of the general meeting of shareholders. The management board is permitted, subject to certain requirements and subject to approval of the supervisory board, to declare interim dividends without the approval of the general meeting of shareholders.
Distributions shall be payable in such currency as determined by our management board. We intend that distributions, if any, shall be payable on such date as determined by our management board. Our management board will set the date that will be applied in order to establish which shareholders (or usufructuaries or pledgees, as the case may be) are entitled to the distribution, such date not being earlier than the date on which the distribution was announced. Claims for payment of dividends and other distributions not made within five years from the date that such dividends or distributions became payable, will lapse and any such amounts will be considered to have been forfeited to us (“verjaring”).
We have never declared or paid any dividends on our ordinary shares, and we currently do not plan to declare dividends on our ordinary shares in the foreseeable future.
B. Significant Changes
There have been no significant changes since the date of the annual financial statements.
Item 9: The Offer and Listing
A. Offering and Listing Details
See “Item 9.C The Offer and Listing - Markets.”
B. Plan of Distribution
Not applicable.
C. Markets
Since September 18, 2014, our ordinary shares have been listed on Nasdaq. Our ordinary shares are currently trading on Nasdaq Capital Market under ticker symbol “PRQR”.
D. Selling Shareholders
Not applicable.
114
E. Dilution
Not applicable.
F. Expenses of the Issue
Not applicable.
Item 10: Additional Information
A. Share Capital
Not applicable.
B. Memorandum and Articles of Association
General
We were incorporated on February 21, 2012 as a private company with limited liability (‘besloten vennootschap met beperkte aansprakelijkheid’) under Dutch law. In connection with our initial public offering in 2014, our shareholders resolved to amend our articles of association and to convert into a public company with limited liability by means of a Deed of Amendment and Conversion, pursuant to which, we converted to a public company with limited liability (‘naamloze vennootschap’) under the laws of the Netherlands. In connection with this conversion, our legal name changed from ProQR Therapeutics B.V. to ProQR Therapeutics N.V. On June 22, 2016 the articles of association were amended to (i) add certain places where general meeting of shareholders may be held and (ii) amend the term ‘annual report’ to ‘report of the Management Board’ to comply with the Implementation Act Annual Accounts Directive (‘Uitvoeringswet richtlijn jaarrekening’) (Bulletin of Acts and Decrees (‘Staatsblad’) 2015, 349), pursuant to which act this term has been amended accordingly. On February 27, 2018, the articles of association were amended to (i) to increase the authorized share capital, and (ii) to delete the requirement of a deed for the issuance of shares. On June 10, 2021, the articles of association were amended to (i) combine the existing compensation committee and nominating and corporate governance committee into one committee, to be named the compensation, nominating and corporate governance committee, and to establish a new research and development committee, and (ii) to increase the authorized share capital.
Our company is registered with the Dutch Trade Register of the Chamber of Commerce (‘handelsregister van de Kamer van Koophandel’) in Leiden, the Netherlands under number 54600790. Our corporate seat is in Leiden, the Netherlands, and our registered office is at Zernikedreef 9, 2333 CK Leiden, the Netherlands.
At December 31, 2023, our authorized share capital is € 13,600,000, divided into 170,000,000 ordinary shares and 170,000,000 preferred shares, each with a nominal value of € 0.04.
Our ordinary shares are listed on the Nasdaq Capital Market under the symbol “PRQR.”
We have listed our ordinary shares in registered form and our shares are not certificated. We have appointed American Stock Transfer & Trust Company, LLC as our agent to maintain our shareholders register and to act as transfer agent, registrar and paying agent for the ordinary shares. Our ordinary shares are traded on the Nasdaq Capital Market in book-entry form.
Articles of Association and Dutch Law
Set forth below is a summary of relevant information concerning the material provisions of our articles of association and applicable Dutch law. This summary does not constitute legal advice regarding those matters and should not be regarded as such.
115
Anti-Takeover Measure
To enable us to pursue our business strategy and the successful development of our product pipeline and to protect our interests and those of our stakeholders (including shareholders, employees and patient populations), our business strategy, our continuity and our independence against actual and potential threats, we have adopted an anti-takeover measure by granting a perpetual and repeatedly exercisable call option to a protection foundation, Stichting Continuity ProQR Therapeutics. The call option confers on the protection foundation the right to acquire under certain conditions such number of preferred shares as equals, at the time of exercise of the call option, the lesser of: (i) the total number of shares equal to our issued share capital at that time, minus the number of preferred shares already held by the protection foundation at that time (if any) or (ii) the maximum number of preferred shares that may be issued under our authorized share capital under our articles of association from time to time. The protection foundation’s articles of association provide that it will act to promote and protect the best interests of us, our business and our stakeholders, including patient populations that may benefit from our products and pipeline, by opposing any influences that conflict with these interests and threaten to undermine our strategy, continuity, independence and identity. The board of the protection foundation is independent from us, our stakeholders and our subsidiaries.
Upon exercise of the call option, the preferred shares will be issued to the protection foundation for their nominal value, of which at least 25% will be due upon issuance, and may also be issued against the Company’s reserves if so requested by the protection foundation. The voting rights of our shares are based on nominal value and as we expect our shares to trade substantially in excess of nominal value, a foundation acquiring preferred shares issued at 25% of their nominal value can obtain significant voting power for a substantially reduced price and thus be used as a defensive measure. These preferred shares will have both a liquidation and dividend preference over our ordinary shares and will accrue cash dividends at a fixed rate with deficits in a preferred dividend being carried forward. The protection foundation may exercise the call option to acquire preferred shares in order to protect us from influences that do not serve our best interests and threaten to undermine our strategy, continuity, independence and identity. These influences may include a third-party acquiring a significant percentage of our ordinary shares, the announcement of a public offer for our ordinary shares, other concentration of control over our ordinary shares or any other form of pressure on us to alter our strategic policies.
Company’s Shareholders’ Register
All of our registered shares are registered in our shareholders’ register. Subject to Dutch law and our articles of association, we must keep our shareholders’ register accurate and up-to-date. Our shareholders’ register shall be kept by our management board. The sub-register shall be kept by our agent on behalf of the management board. Our shareholders’ register includes the names and addresses and other relevant details of all holders of registered shares, and shows the date on which the shares were acquired, the date of the acknowledgement by, or notification of, us as well as the amount paid on each share. The shareholders’ register also includes the names and addresses and other relevant details of those with a right of usufruct (‘vruchtgebruik’) or a right of pledge (‘pandrecht’) in respect of any shares. Our registered ordinary shares are held through DTC and therefore DTC is recorded in the shareholders register as the holder of those ordinary shares.
Shareholders, usufructuaries and pledgees whose particulars must be recorded in our shareholders’ register are required to provide our management board with the necessary particulars in a timely fashion. Upon request, shareholders, usufructuaries and pledgees shall be provided with an extract of our shareholders’ register in respect of their right to one or more registered shares.
116
Issuance of Shares and Pre-emptive Rights
Under Dutch law, shares are issued and rights to subscribe for shares are granted pursuant to a resolution of the general meeting of shareholders. Our articles of association provide that our general meeting of shareholders may only adopt such resolution upon a proposal of our management board, which proposal must have been approved by our supervisory board. Our general meeting of shareholders may authorize our management board to issue new shares or grant rights to subscribe for shares. The authorization can be granted and extended, in each case for a period not exceeding five years. For as long as such authorization is effective, our general meeting of shareholders will not have the power to issue shares and rights to subscribe for shares. Pursuant to our articles of association, our management board may only exercise the power to issue shares with the approval of our supervisory board.
Under Dutch law, in the event of an issuance of ordinary shares or granting of rights to subscribe for ordinary shares, each shareholder will have a pro rata preemptive right in proportion to the aggregate nominal value of the ordinary shares held by such holder. A holder of ordinary shares does not have a preemptive right with respect to the issuance of, or granting of rights to subscribe for, (i) ordinary shares for consideration other than cash, or (ii) ordinary shares to our employees or employees of one of our group companies, or (iii) ordinary shares to persons exercising a previously granted right to subscribe for shares, or (iv) preferred shares.
The preemptive rights in respect of newly issued ordinary shares may be restricted or excluded by a resolution of the general meeting of shareholders. Our articles of association provide that our general meeting of shareholders may only adopt such resolution upon a proposal of our management board, which proposal must have been approved by our supervisory board. Our general meeting of shareholders may authorize our management board to restrict or exclude the preemptive rights in respect of newly issued ordinary shares. Such authorization for the management board can be granted and extended, in each case for a period not exceeding five years. For as long as such authorization is effective, our general meeting of shareholders will not have the power to limit or exclude preemptive rights and such authorization may not be revoked unless stipulated otherwise in the authorization. A resolution of the general meeting of shareholders to restrict or exclude the preemptive rights or to designate our management board as the authorized body to do so requires at least a two-thirds majority of the votes cast, if less than one half of our issued share capital is represented at the meeting.
Preferred shares do not carry preemptive rights in respect of newly issued ordinary shares or preferred shares, nor do holders of ordinary shares have preemptive rights in respect of newly issued preferred shares. The call option of the protection foundation to acquire newly issued preferred shares of the company, see “Description of Share Capital—Anti-Takeover Measure”, is an irrevocable and repeatedly exercisable right to subscribe for preferred shares.
On September 15, 2014, our general meeting of shareholders adopted a resolution pursuant to which our management board will be irrevocably authorized to, following approval of our supervisory board, limit or exclude the preemptive rights of holders of ordinary shares for a period of five years from the date of such resolution.
On May 16, 2018, our general meeting of shareholders adopted a resolution pursuant to which the aforesaid authorizations to issue shares and to limit and exclude preemptive rights was renewed. In this renewed authorization the Management Board was delegated, subject to approval of the Supervisory Board, the authority for a period of 5 years from the date of the resolution of the general meeting of shareholders to, in accordance with applicable laws and Nasdaq listing rules: (a) issue an amount of ordinary shares up to 100% of the Company's authorized share capital for general purposes as reflected above and issuances under Company’s stock option plans with the provision that the issuances under stock option plans is limited to 15% of the Company's issued share capital (minus any treasury shares) at May 16, 2018; (b) grant rights to subscribe for ordinary shares as described under (a); and (c) limit or exclude the pre-emptive rights of holders of ordinary shares, which delegation shall include the authority to determine the price and further terms and conditions of any such share issuance or grants.
117
On June 23, 2020, our general meeting of shareholders adopted a resolution pursuant to which the aforesaid authorizations to issue shares and to limit and exclude preemptive rights was renewed. In this renewed authorization the Management Board was delegated, subject to approval of the Supervisory Board, the authority for a period of 5 years from the date of the resolution of the general meeting of shareholders to, in accordance with applicable laws and Nasdaq listing rules: (a) issue an amount of ordinary shares up to 100% of the Company's authorized share capital for general purposes as reflected above and issuances under Company’s equity incentive or stock option plans with the provision that the issuances under equity incentive or stock option plans are limited to 15% of the Company's issued share capital from time-to-time (minus any treasury shares); (b) grant rights to subscribe for ordinary shares as described under (a); and (c) limit or exclude the pre-emptive rights of holders of ordinary shares, which delegation shall include the authority to determine the price and further terms and conditions of any such share issuance or grant.
On May 19, 2021, our general meeting of shareholders adopted a resolution pursuant to which the aforesaid authorizations to issue shares and to limit and exclude preemptive rights was renewed. In this renewed authorization the Management Board was delegated, subject to approval of the Supervisory Board, the authority for a period of 5 years from the date of the resolution of the general meeting of shareholders to, in accordance with applicable laws and Nasdaq listing rules: (a) issue ordinary shares up to 100% of the Company’s authorized share capital for general purposes and issuances under Company’s equity incentive or stock option plans with the provision that the issuances under equity incentive or stock option plans is limited to 15% of the Company’s issued share capital from time-to-time (minus any treasury shares); (b) grant rights to subscribe for ordinary shares as described under (a); and (c) limit or exclude the pre-emptive rights of holders of ordinary shares, which delegation shall include the authority to determine the price and further terms and conditions of any such share issuance or grant.
On June 30, 2022, our general meeting of shareholders adopted a resolution pursuant to which the aforesaid authorizations to issue shares and to limit and exclude preemptive rights was renewed. In this renewed authorization the Management Board was delegated, subject to approval of the Supervisory Board, the authority for a period of 5 years from the date of the resolution of the general meeting of shareholders to, in accordance with applicable laws and Nasdaq listing rules: (a) issue ordinary shares up to 100% of the Company’s authorized share capital for general purposes and issuances under Company’s equity incentive or stock option plans with the provision that the issuances under equity incentive or stock option plans is limited to 15% of the Company’s issued share capital from time-to-time (minus any treasury shares); (b) grant rights to subscribe for ordinary shares as described under (a); and (c) limit or exclude the pre-emptive rights of holders of ordinary shares, which delegation shall include the authority to determine the price and further terms and conditions of any such share issuance or grant.
On May 17, 2023, our general meeting of shareholders adopted a resolution pursuant to which the aforesaid authorizations to issue shares and to limit and exclude preemptive rights was renewed. In this renewed authorization the Management Board was delegated, subject to approval of the Supervisory Board, the authority for a period of 5 years from the date of the resolution of the general meeting of shareholders to, in accordance with applicable laws and Nasdaq listing rules: (a) issue ordinary shares up to 100% of the Company’s authorized share capital for general purposes and issuances under Company’s equity incentive or stock option plans with the provision that the issuances under equity incentive or stock option plans is limited to 15% of the Company’s issued share capital from time-to-time (minus any treasury shares); (b) grant rights to subscribe for ordinary shares as described under (a); and (c) limit or exclude the pre-emptive rights of holders of ordinary shares, which delegation shall include the authority to determine the price and further terms and conditions of any such share issuance or grant.
Repurchases of our Shares
Under Dutch law, we may not subscribe for newly issued shares in our own capital. We may acquire our shares, subject to applicable provisions and restrictions of Dutch law and our articles of association, to the extent that:
| ● | such shares are fully paid-up; |
| ● | such shares are acquired for no consideration or such repurchase would not cause our shareholders’ equity to fall below an amount equal to the sum of the paid-up and called-up part of the issued share capital and the reserves we are required to maintain pursuant to Dutch law or our articles of association; and |
118
| ● | after the acquisition of shares, we and our subsidiaries would not hold, or would not hold as pledgees, shares having an aggregate nominal value that exceeds 50% of our issued share capital. |
Other than shares acquired for no consideration or by universal succession, we may acquire shares only if our general meeting of shareholders has authorized the management board to do so. An authorization by the general meeting of shareholders for the acquisition of shares can be granted for a maximum period of 18 months. Such authorization must specify the number of shares that may be acquired, the manner in which these shares may be acquired and the price range within which the shares may be acquired. No authorization of the general meeting of shareholders is required if ordinary shares are acquired by us on Nasdaq with the intention of transferring such ordinary shares to our employees or employees of a group company pursuant to an arrangement applicable to them. Our articles of association further provide that a resolution of our management board to acquire fully paid-up shares in our share capital requires the approval of our supervisory board.
On May 10, 2017, our general meeting of shareholders adopted a resolution pursuant to which our management board is authorized to acquire up to 10% of our issued share capital plus, in case of a material reorganization of the capital structure of the Company an additional 10% on Nasdaq or by other means for an 18 month period from the date of such resolution for a price per share not exceeding 110% of the market price of the ordinary shares on Nasdaq (with the market price deemed to be the average of the closing price on each of the five consecutive days of trading preceding the three trading days prior to the date of acquisition).
On June 23, 2020 our general meeting of shareholders adopted a resolution pursuant to which the aforementioned authorization to acquire shares was renewed. In this renewed authorization the Management Board was delegated the authority to perform acquisitions by the Company of (i) up to 10% of the issued share capital of the Company plus, (ii) in case of a material reorganization of the capital structure of the Company, an additional 10% of the issued share capital of the Company, by any means, including through derivative products, purchases on any stock exchange, through any private purchase or block trade, or otherwise, for a price that is between 0.01 US Dollar and an amount which is not higher than 110% of the average market price of such ordinary shares on Nasdaq (with the market price deemed to be the average of the closing price on each of the five consecutive days of trading preceding the three trading days prior to the date of acquisition), for a period of 18 months with effect from the AGM.
On May 19, 2021, our general meeting of shareholders adopted a resolution pursuant to which the aforementioned authorization to acquire shares was renewed. In this renewed authorization the Management Board was delegated the authority to perform acquisitions by the Company of (i) up to 10% of the issued share capital of the Company plus, in case of a material reorganization of the capital structure of the Company, (ii) an additional 10% of the issued share capital of the Company, by any means, including through derivative products, purchases on any stock exchange, through any private purchase or block trade, or otherwise, for a price that is between 0.01 US Dollar and an amount which is not higher than 110% of the average market price of such ordinary shares on Nasdaq (with the market price deemed to be the average of the closing price on each of the five consecutive days of trading preceding the three trading days prior to the date of acquisition), for a period of eighteen (18) months with effect from the general meeting of shareholders. In this respect, the words “issued share capital” means the Company’s issued share capital from time to time. For the avoidance of doubt, the issued share capital includes treasury shares.
On June 30, 2022, our general meeting of shareholders adopted a resolution pursuant to which the aforementioned authorization to acquire shares was renewed. In this renewed authorization the Management Board was delegated the authority to perform acquisitions by the Company of (i) up to 10% of the issued share capital of the Company plus, in case of a material reorganization of the capital structure of the Company, (ii) an additional 10% of the issued share capital of the Company, by any means, including through derivative products, purchases on any stock exchange, through any private purchase or block trade, or otherwise, for a price that is between 0.01 US Dollar and an amount which is not higher than 110% of the average market price of such ordinary shares on Nasdaq (with the market price deemed to be the average of the closing price on each of the five consecutive days of trading preceding the three trading days prior to the date of acquisition), for a period of eighteen (18) months with effect from the general meeting of shareholders. In this respect, the words “issued share capital” means the Company’s issued share capital from time to time. For the avoidance of doubt, the issued share capital includes treasury shares.
119
On May 17, 2023, our general meeting of shareholders adopted a resolution pursuant to which the aforementioned authorization to acquire shares was renewed. In this renewed authorization the Management Board was delegated the authority to perform acquisitions by the Company of (i) up to 10% of the issued share capital of the Company plus, in case of a material reorganization of the capital structure of the Company, (ii) an additional 10% of the issued share capital of the Company, by any means, including through derivative products, purchases on any stock exchange, through any private purchase or block trade, or otherwise, for a price that is between 0.01 US Dollar and an amount which is not higher than 110% of the average market price of such ordinary shares on Nasdaq (with the market price deemed to be the average of the closing price on each of the five consecutive days of trading preceding the three trading days prior to the date of acquisition), for a period of eighteen (18) months with effect from the general meeting of shareholders. In this respect, the words “issued share capital” means the Company’s issued share capital from time to time. For the avoidance of doubt, the issued share capital includes treasury shares.
Capital Reductions; Cancellation
At a general meeting of shareholders, our shareholders may resolve to reduce our issued share capital by (i) cancelling shares or (ii) reducing the nominal value of the shares by virtue of an amendment to our articles of association. In either case, this reduction would be subject to applicable statutory provisions. A resolution to cancel shares may only relate (x) to shares held by the company itself or in respect of which the company holds the depository receipts, and (y) to all preferred shares. Our articles of association provide that our general meeting of shareholders may only adopt such a resolution upon a proposal of our management board, which proposal must have been approved by our supervisory board. In order to be adopted by the general meeting of shareholders, a resolution to reduce the capital requires a simple majority of the votes cast at a general meeting of shareholders if at least half the issued capital is represented at the meeting or at least two-thirds of the votes cast at the general meeting of shareholders if less than half of the issued capital is represented at the general meeting of shareholders.
A reduction of the nominal value of shares without repayment and without release from the obligation to pay up the shares must be effectuated proportionally on shares of the same class (unless all shareholders concerned agree to a disproportionate reduction). A resolution that would result in a reduction of capital requires approval of the meeting of each group of holders of shares of the class or classes whose rights are prejudiced by the reduction. In addition, a reduction of capital involves a two-month waiting period during which creditors have the right to object to a reduction of capital under specified circumstances.
In the event that all preferred shares are cancelled, distributions shall be made to the protection foundation as sole holder of such preferred shares.
Corporate Objectives
Under our articles of association, our corporate objectives are:
| ● | the development, bringing to market and sale of products and technologies in the field of biotechnology; |
| ● | the research and development of (or the commission to research and develop) patents, know-how and intellectual and industrial property; |
| ● | to make our products available to the patient populations that may benefit from such products and to maintain a suitable pipeline of products that may be beneficial for relevant patient populations; |
| ● | to participate in, to finance, to hold any other interest in and to conduct the management or supervision of other entities, companies, partnerships and businesses; |
| ● | to furnish guarantees, to provide security, to warrant performance in any other way and to assume liability, whether jointly and severally or otherwise, in respect of obligations of group companies or other parties; and |
120
| ● | to do anything which, in the widest sense of the words, is connected with or may be conducive to the attainment of these objects. |
Amendment of Articles of Association
Our general meeting of shareholders, at the proposal of our management board, with the prior approval of our supervisory board, may resolve to amend our articles of association. A resolution taken by the general meeting of shareholders to amend our articles of association requires a simple majority of the votes cast.
General Meetings of Shareholders
General meetings of shareholders can be held in Leiden, Amsterdam, Rotterdam, Schiphol Airport (municipality Haarlemmermeer), The Hague, Oegstgeest, Leidschendam, Katwijk, Noordwijk or Wassenaar, the Netherlands. All shareholders and others entitled to attend general meetings of shareholders are authorized to attend the general meeting of shareholders, to address the meeting and, in so far as they have such right, to vote, either in person or by proxy.
We must hold at least one general meeting of shareholders each year, to be held within six months after the end of our financial year. A general meeting of shareholders shall also be held within three months after our management board has considered it to be likely that the company’s equity has decreased to an amount equal to or lower than half of its paid up and called up capital. If the management board and supervisory board have failed to ensure that such general meetings of shareholders as referred to in the preceding sentences are held in a timely fashion, each shareholder and other person entitled to attend shareholders’ meetings may be authorized by the Dutch court to convene the general meeting of shareholders.
Our management board and our supervisory board may convene additional extraordinary general meetings of shareholders whenever they so decide. Pursuant to Dutch law, one or more shareholders and/or others entitled to attend general meetings of shareholders, alone or jointly representing at least ten percent of our issued share capital may on their application, be authorized by the Dutch court to convene a general meeting of shareholders. The Dutch court will disallow the application if it does not appear to it that the applicants have previously requested that the management board or supervisory board convenes a shareholders’ meeting and neither the management board nor the supervisory board has taken the necessary steps so that the shareholders’ meeting could be held within eight weeks after the request.
General meetings of shareholders are convened by a notice which includes an agenda stating the items to be discussed. For the annual general meeting of shareholders the agenda will include, among other things, the adoption of our annual accounts, the appropriation of our profits or losses and proposals relating to the composition and filling of any vacancies of the management board or supervisory board. In addition, the agenda for a general meeting of shareholders may include such items as have been included therein by our management board or our supervisory board. Pursuant to Dutch law, one or more shareholders and/or others entitled to attend general meetings of shareholders, alone or jointly representing at least 3% of the issued share capital have the right to request the inclusion of additional items on the agenda of shareholders’ meetings. Such requests must be made in writing, substantiated, or by a proposal for a resolution and received by us no later than the 60th day before the day that the relevant general meeting of shareholders is to be held. No resolutions will be adopted on items other than those which have been included in the agenda.
We will give notice of each general meeting of shareholders by publication on our website and, to the extent required by applicable law, in a Dutch daily newspaper with national distribution, and in any other manner that we may be required to follow in order to comply with Dutch law and applicable stock exchange and SEC requirements. We will observe the statutory minimum convening notice period for a general meeting of shareholders.
121
Pursuant to our articles of association, our management board may determine a record date (‘registratiedatum’) of 28 calendar days prior to a general meeting of shareholders to establish which shareholders and others with meeting rights are entitled to attend and, if applicable, vote in the general meeting of shareholders. The record date, if any, and the manner in which shareholders can register and exercise their rights will be set out in the convocation notice of the general meeting of shareholders. Our articles of association provide that a shareholder must notify the company in writing of his identity and his intention to attend (or be represented at) the general meeting of shareholders, such notice to be received by us ultimately on the seventh day prior to the general meeting of shareholders. If this requirement is not complied with or if upon direction of the company to that effect no proper identification is provided by any person wishing to enter the general meeting of shareholders, the chairperson of the general meeting of shareholders may, in his or her sole discretion, refuse entry to the shareholder or his proxy holder.
Pursuant to our articles of association, our general meeting of shareholders is chaired by the chairman of our supervisory board. If the chairman of our supervisory board is absent and has not charged another person to chair the meeting in his place, the supervisory board members present at the meeting shall appoint one of themselves to be chairperson. If no supervisory board members are present at the general meeting of shareholders, the general meeting of shareholders will be chaired by our CEO or, if our CEO is absent, another management board member present at the meeting and, if none of them is present, the general meeting of shareholders shall appoint its own chairperson. The person who should chair the meeting may appoint another person in his or her stead.
The chairperson of the general meeting of shareholders may decide at his or her discretion to admit other persons to the meeting. The chairperson of the general meeting of shareholders shall appoint another person present at the shareholders’ meeting to act as secretary and to minute the proceedings at the meeting. The chairperson of the general meeting of shareholders may instruct a civil law notary to draw up a notarial report of the proceedings at the company’s expense, in which case no minutes need to be taken. The chairperson of the general meeting of shareholders is authorized to eject any person from the general meeting of shareholders if the chairperson considers that person to disrupt the orderly proceedings. The general meeting of shareholders shall be conducted in the English language.
Voting Rights and Quorum Requirements
In accordance with Dutch law and our articles of association, each issued ordinary share and preferred share confers the right on the holder thereof to cast one vote at the general meeting of shareholders. The voting rights attached to any shares held by us or our direct or indirect subsidiaries are suspended as long as they are held in treasury. Dutch law does not permit cumulative voting for the election of management board members or supervisory board members.
Voting rights may be exercised by shareholders or by a duly appointed proxy holder (the written proxy being acceptable to the chairperson of the general meeting of shareholders) of a shareholder, which proxy holder need not be a shareholder. Our articles of association do not limit the number of shares that may be voted by a single shareholder.
Under our articles of association, blank votes, abstentions and invalid votes shall not be counted as votes cast. Further, shares in respect of which a blank or invalid vote has been cast and shares in respect of which the person with meeting rights who is present or represented at the meeting has abstained from voting are counted when determining the part of the issued share capital that is present or represented at a general meeting of shareholders. The chairperson of the general meeting of shareholders shall determine the manner of voting and whether voting may take place by acclamation.
In accordance with Dutch law and generally accepted business practices, our articles of association do not provide quorum requirements generally applicable to general meetings of shareholders. To this extent, our practice varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting shares.
Resolutions of the general meeting of shareholders are adopted by a simple majority of votes cast without quorum requirement, except where Dutch law or our articles of association provide for a special majority and/or quorum in relation to specified resolutions.
122
The chairperson of the general meeting of shareholders shall decide on the method of voting and may determine the voting procedure. The determination made by the chairperson of the general meeting of shareholders with regard to the results of a vote shall be decisive. However, where the accuracy of the chairperson’s determination is contested immediately after it has been made, a new vote shall take place if the majority of the general meeting of shareholders so requires or, where the original vote did not take place by response to a roll call or in writing, if any party with voting rights present at the meeting so requires.
Our management board will keep a record of the resolutions passed at each general meeting of shareholders. The record shall be available at our office for inspection by any person entitled to attend general meetings of shareholders and upon request a copy of or extract from the record will be provided to such person at no more than the cost price.
Our articles of association and Dutch law provide that resolutions of our management board concerning a material change in the identity or character of the company or our business are subject to the approval of the general meeting of shareholders. Such changes include in any event:
| ● | a transfer of all or materially all of our business to a third party; |
| ● | the entry into or termination of a long-lasting alliance of the company or of a subsidiary either with another entity or company, or as a fully liable partner of a limited partnership or partnership, if this alliance or termination is of significant importance for the company; and |
| ● | the acquisition or disposition of an interest in the capital of a company by the company or by a subsidiary with a value of at least one third of the value of the assets, according to the balance sheet with explanatory notes or, if the company prepares a consolidated balance sheet, according to the consolidated balance sheet with explanatory notes in the company’s most recently adopted annual accounts. |
Adoption of Annual Accounts and Discharge of Management and Supervisory Liability
Pursuant to Dutch law, we are required to publish our annual accounts within eight days after adoption and ultimately within 13 months after the end of our financial year.
Each year within five months after the end of our financial year, save where this period is extended for a maximum of five months by the general meeting of shareholders on account of special circumstances, our management board will prepare the annual accounts. The annual accounts must be accompanied by an auditor’s certificate, a report of the management board and certain other mandatory information and must be made available for inspection by our shareholders at our offices within the same period. Under Dutch law, the general meeting of shareholders may appoint and remove our independent auditors, as referred to in Section 2:393 Dutch Civil Code, who audit the annual accounts. If the general meeting of shareholders fails to appoint an independent auditor, the auditor will be appointed by the supervisory board or, if the supervisory board fails to do so, the management board. The annual accounts are adopted by our shareholders at the general meeting of shareholders and will be prepared in accordance with Part 9 of Book 2 of the Dutch Civil Code.
The adoption of the annual accounts by our shareholders does not release our management board members and our supervisory board members from liability for acts reflected in those documents. Any such release from liability requires a separate shareholders’ resolution.
Our financial reporting will be subject to the supervision of the Dutch regulator AFM. The AFM will review the content of the financial reports and has the authority to approach us with requests for information if, on the basis of publicly available information, it has reasonable doubts as to the integrity of our financial reporting. For a more detailed description we refer to the description below under the heading “Dutch Financial Reporting Supervision Act.”
123
Dividends and other Distributions
We may only make distributions to our shareholders and other persons entitled to distributable profits, to the extent that our shareholders’ equity exceeds the sum of the paid-up and called-up share capital plus the reserves as required to be maintained by Dutch law or by our articles of association.
Under our articles of association, a (cumulative) dividend is first paid out of the profit, if available for distribution, on any preferred shares. Any amount remaining out of the profit is carried to reserve as the management board determines. After reservation by the management board of any profit, the remaining profit will be at the disposal of the general meeting of shareholders. The management board is permitted, subject to certain requirements and subject to approval of the supervisory board, to declare interim dividends without the approval of the general meeting of shareholders.
Distributions shall be payable in such currency as determined by our management board. We intend that distributions, if any, shall be payable on such date as determined by our management board. Our management board will set the date that will be applied in order to establish which shareholders (or usufructuaries or pledgees, as the case may be) are entitled to the distribution, such date not being earlier than the date on which the distribution was announced. Claims for payment of dividends and other distributions not made within five years from the date that such dividends or distributions became payable, will lapse and any such amounts will be considered to have been forfeited to us (‘verjaring’).
We do not anticipate paying any dividends for the foreseeable future.
Liquidation and Dissolution
The general meeting of shareholders may, based on a proposal by our management board, which proposal has been approved by our supervisory board, resolve to dissolve the company by a resolution passed by a simple majority of the votes cast. In the event of the company being dissolved, the liquidation shall be effected by our management board under the supervision of the supervisory board, unless the general meeting of shareholders decides otherwise.
In the event of a dissolution and liquidation, the assets remaining after payment of all of the company’s debts (including any liquidation expenses) are to be distributed (i) firstly to the holders, if any, of preferred shares in the amount of the nominal value of the preferred shares (to the extent paid-up) plus unpaid accrued dividends and deficits (if any) in preferred dividends, and (ii) the balance remaining to the holders of ordinary shares in proportion to the aggregate nominal value of their ordinary shares. The liquidation and all distributions referred to in this paragraph will be made in accordance with the relevant provisions of Dutch law.
Limitations on Non-Residents and Exchange Controls
There are no limits under the laws of the Netherlands or in our articles of association on non-residents of the Netherlands holding or voting our ordinary shares. Under Dutch law, there are currently no exchange controls applicable to the transfer of dividends or other distributions with respect to, or of the proceeds from the sale of, shares in a Dutch company, to persons outside the Netherlands.
124
Netherlands Squeeze-Out Proceedings
Pursuant to Section 2:92a of the Dutch Civil Code, a shareholder who for its own account (or together with its group companies) holds at least 95% of our issued share capital may institute proceedings against our other shareholders jointly for the transfer of their shares to it. The proceedings are held before the Enterprise Chamber of the Amsterdam Court of Appeal (‘Ondernemingskamer’) and can be instituted by means of a writ of summons served upon each of the minority shareholders in accordance with the provisions of the Dutch Code of Civil Procedure (‘Wetboek van Burgerlijke Rechtsvordering’). The Enterprise Chamber may grant the claim for squeeze-out in relation to all minority shareholders and will determine the price to be paid for the shares, if necessary after appointment of one or three experts who will offer an opinion to the Enterprise Chamber on the value of the shares of the minority shareholders. Once the order to transfer by the Enterprise Chamber of the Amsterdam Court of Appeal becomes final and irrevocable, the majority shareholder that instituted the squeeze-out proceedings shall give written notice of the date and place of payment and the price to the holders of the shares to be acquired whose addresses are known to the majority shareholder. Unless the addresses of all minority shareholders are known to the majority shareholder acquiring the shares, the majority shareholder is required to publish the same notice in a newspaper with a national circulation.
A shareholder that holds a majority of our issued share capital, but less than the 95% required to institute the squeeze-out proceedings described above, may seek to propose and implement one or more restructuring transactions with the objective to obtain at least 95% of our issued share capital and thus to be allowed to initiate squeeze-out proceedings. Those restructuring transactions could, amongst other things, include a legal merger or demerger involving our company, a contribution of cash and/or assets against issuance of shares involving our company, and the issue of new shares to the majority shareholder while excluding any pre-emption rights of minority shareholders in relation to such issuance or an asset sale transaction.
In Dutch public takeover practice, depending on the circumstances, an asset sale transaction is sometimes used as a way to squeeze out minority shareholders (e.g. after a successful public offer, or tender offer, through which the offeror acquires a supermajority, but less than all, of the shares). In such a scenario, the business of the target company would be sold to an offeror, a buyer or a special purpose vehicle, followed by the liquidation of the target company. The purchase price would be distributed to all shareholders in proportion to their respective shareholding as liquidation proceeds, thus separating the business from the company in which minority shareholders participated.
Under our articles of association, any proposal to sell and transfer all of our assets and to dissolve and liquidate our company is subject to approval by a majority of the votes cast in our general meeting of shareholders which must be preceded by a proposal by our management board, which must be approved by our supervisory board.
C. Material Contracts
Except as otherwise disclosed in this Annual Report (including the Exhibits), we are currently not, and have not been in the two years preceding publication of this Annual Report, party to any material contract, other than contracts entered into in the ordinary course of business.
D. Exchange Controls
Cash dividends payable on our ordinary shares may be officially transferred from the Netherlands to non-residents and converted into any other currency without legal restrictions imposed by Dutch law, except that for statistical purposes such payments and transactions must be reported to the Dutch Central Bank. Furthermore, no payments, including dividend payments, may be made to jurisdictions subject to sanctions adopted by the government of the Netherlands and implementing resolutions of the Security Council of the United Nations.
125
E. Taxation
Taxation in the Netherlands
General
The following is a general summary of certain material Dutch tax consequences of the holding and disposal of the ordinary shares. This summary does not purport to describe all possible tax considerations or consequences that may be relevant to all categories of investors, some of which may be subject to special treatment under applicable law (such as trusts or other similar arrangements), and in view of its general nature, it should be treated with corresponding caution. Holders should consult with their tax advisors with regard to the tax consequences of investing in the ordinary shares in their particular circumstances. The discussion below is included for general information purposes only.
Please note that this summary does not describe the tax considerations for:
| (i) | holders of ordinary shares if such holders, and in the case of individuals, his/her partner or certain of their relatives by blood or marriage in the direct line (including foster children), have a substantial interest or deemed substantial interest in us under the Dutch Income Tax Act 2001 (in Dutch: ‘Wet inkomstenbelasting 2001’). Generally speaking, a holder of securities in a company is considered to hold a substantial interest in such company, if such holder alone or, in the case of individuals, together with his/her partner (statutorily defined term), directly or indirectly, holds (i) an interest of 5% or more of the total issued and outstanding capital of that company or of 5% or more of the issued and outstanding capital of a certain class of shares of that company; or (ii) rights to acquire, directly or indirectly, such interest; or (iii) certain profit sharing rights in that company that relate to 5% or more of the company’s annual profits and/or to 5% or more of the company’s liquidation proceeds. A deemed substantial interest may arise if a substantial interest (or part thereof) in a company has been disposed of, or is deemed to have been disposed of, on a non-recognition basis; |
| (ii) | holders of ordinary shares in us that qualify or qualified as a participation for purposes of the Dutch Corporate Income Tax Act 1969 (in Dutch: ‘Wet op de vennootschapsbelasting 1969’). Generally, a taxpayer’s shareholding of 5% or more in a company’s nominal paid-up share capital qualifies as a participation. A holder may also have a participation if such holder does not have a 5% shareholding but a related entity (statutorily defined term) has a participation or if the company in which the shares are held is a related entity (statutorily defined term); |
| (iii) | holders of ordinary shares who are individuals for whom the ordinary shares or any benefit derived from the ordinary shares are compensation or deemed to be compensation for employment activities, including deemed employment activities performed by such holders or certain individuals related to such holders (as defined in the Dutch Income Tax Act 2001) or statutory directors (‘bestuurders’) or supervisory directors (‘commissarissen’) of a company resident in the Netherlands; and |
| (iv) | pension funds, investment institutions (in Dutch: ‘fiscale beleggingsinstellingen’), exempt investment institutions (in Dutch: ‘vrijgestelde beleggingsinstellingen’) and other entities that are, in whole or in part, not subject to or exempt from corporate income tax in the Netherlands, as well as entities that are exempt from corporate income tax in their country of residence, such country of residence being another state of the European Union, Norway, Liechtenstein, Iceland or any other state with which the Netherlands have agreed to exchange information in line with international standards. |
Except as otherwise indicated, this summary only addresses national tax legislation and published regulations in the Netherlands, whereby the Netherlands means the part of the Kingdom of the Netherlands located in Europe, as in effect on the date hereof and as interpreted in published case law until this date, without prejudice to any amendment introduced at a later date and implemented with or without retroactive effect.
126
(a) Dividend Withholding Tax
Dividends distributed by us generally are subject to Dutch dividend withholding tax at a rate of 15%. The expression “dividends distributed” includes, among other things:
| ● | distributions in cash or in kind, deemed and constructive distributions and repayments of paid-in capital not recognized for Dutch dividend withholding tax purposes; |
| ● | liquidation proceeds, proceeds of redemption of ordinary shares, or proceeds of the repurchase of ordinary shares by us or one of our subsidiaries to the extent such proceeds exceed the average paid-in capital of those shares as recognized for purposes of Dutch dividend withholding tax; |
| ● | an amount equal to the par value of ordinary shares issued or an increase of the par value of ordinary shares, to the extent that it does not appear that a contribution, recognized for purposes of Dutch dividend withholding tax, has been made or will be made; and |
| ● | partial repayment of the paid-in capital, recognized for purposes of Dutch dividend withholding tax, if and to the extent that we have net profits (in Dutch: ‘zuivere winst’), unless the holders of ordinary shares have resolved in advance at a general meeting to make such repayment and the par value of the ordinary shares concerned has been reduced by an equal amount by way of an amendment of our articles of association. |
If a holder of ordinary shares is resident in a country other than the Netherlands and if a double taxation convention is in effect between the Netherlands and such other country, such holder of ordinary shares may, depending on the terms of that double taxation convention, be eligible for a full or partial exemption from, or refund of, Dutch dividend withholding tax.
Individuals and corporate legal entities who are resident or deemed to be resident in the Netherlands for Dutch tax purposes (‘Dutch Resident Individuals’ and ‘Dutch Resident Entities’ as the case may be), can generally credit the Dutch dividend withholding tax against their income tax or corporate income tax liability. The same generally applies to holders of ordinary shares that are neither resident nor deemed to be resident of the Netherlands if the ordinary shares are attributable to a Dutch permanent establishment of such non-resident holder.
In general, we will be required to remit all amounts withheld as Dutch dividend withholding tax to the Dutch tax authorities. However, under certain circumstances, we are allowed to reduce the amount to be remitted to the Dutch tax authorities by the lesser of:
| ● | 3% of the portion of the distribution paid by us that is subject to Dutch dividend withholding tax; and |
| ● | 3% of the dividends and profit distributions, before deduction of foreign withholding taxes, received by us from qualifying foreign subsidiaries in the current calendar year (up to the date of the distribution by us) and the two preceding calendar years, as far as such dividends and profit distributions have not yet been taken into account for purposes of establishing the above mentioned reduction. (Qualifying foreign subsidiaries are entities established in Aruba, Curacao, St. Maarten, the BES islands or in a state which has concluded a double tax treaty with the Netherlands) |
Although this reduction reduces the amount of Dutch dividend withholding tax that we are required to remit to the Dutch tax authorities, it does not reduce the amount of tax that we are required to withhold on dividends distributed.
127
Pursuant to legislation to counteract “dividend stripping”, a reduction, exemption, credit or refund of Dutch dividend withholding tax is denied if the recipient of the dividend is not the beneficial owner as described in the Dutch Dividend Withholding Tax Act 1965 (in Dutch: ‘Wet op de dividendbelasting 1965’). This legislation generally targets situations in which a shareholder retains its economic interest in shares but reduces the withholding tax costs on dividends by a transaction with another party. It is not required for these rules to apply that the recipient of the dividends is aware that a dividend stripping transaction took place. The Dutch State Secretary of Finance takes the position that the definition of beneficial ownership introduced by this legislation will also be applied in the context of a double taxation convention.
(b) Taxes on Income and Capital Gains
(i) Dutch Resident Individuals
If a holder of ordinary shares is a Dutch Resident Individual, any benefit derived or deemed to be derived from the ordinary shares is taxable at the progressive income tax rates (with a maximum of 49.50%), if:
| (a) | the ordinary shares are attributable to an enterprise from which the Dutch Resident Individual derives a share of the profit, whether as an entrepreneur or as a person who has a co-entitlement to the net worth (in Dutch: ‘medegerechtigd tot het vermogen’) of such enterprise, without being an entrepreneur or a shareholder, as defined in the Dutch Income Tax Act 2001; or |
| (b) | the holder of the ordinary shares is considered to perform activities with respect to the ordinary shares that go beyond ordinary asset management (in Dutch: ‘normaal, actief vermogensbeheer’) or derives benefits from the ordinary shares that are taxable as benefits from other activities (in Dutch ‘resultaat uit overige werkzaamheden’). |
If the above-mentioned conditions (a) and (b) do not apply to the individual holder of ordinary shares, the ordinary shares are recognized as investment assets and included as such in such holder’s net investment asset base (in Dutch: ‘rendementsgrondslag’, ‘Box 3’). Box 3 consists of income from savings and (passive) investments. The value of the box 3 assets per January 1st of each calendar year is, in short, deemed to yield income at a rate which depends on the type of asset: savings (0.92%), investments/other assets (6.17%) or debts (2.46%). Since this Box 3 tax is based on a deemed income, the actual return on the investments is not relevant. The deemed income is taxed at a flat rate of 32% (2023). Taxation only occurs if and to the extent that the fair market value of the assets reduced by the liabilities exceeds a threshold (in Dutch: ‘heffingsvrij vermogen’) of € 57,000 (or € 114,000 in case of a fiscal partnership). The deemed return will be adjusted annually.
On December 24, 2021, the Supreme Court of the Netherlands ruled in a class-action appeal that the Box 3 tax regime for the years 2017 and 2018 was in conflict with the European Convention on Human Rights (“ECHR”), specifically the right to uninterrupted enjoyment of property and the prohibition on discrimination. In addition, the Supreme Court ruled that not the deemed return, but (only) the actual return should be taxed. The judgement affected the years from 2017 onwards and led to the current (temporary) Box 3 legislation.
It was announced that as of 2027 a new Box 3 regime will be implemented. Nevertheless, at this moment, there is still some uncertainty about (the future of) Box 3, as the legislative proposal is still in draft.
(ii) Dutch Resident Entities
Any benefit derived or deemed to be derived from the ordinary shares held by Dutch Resident Entities, including any capital gains realized on the disposal thereof, will generally be subject to Dutch corporate income tax at a rate of 25.8% (a corporate income tax rate of 19% applies with respect to taxable profits up to € 200,000) (2023).
128
(iii) Non-Residents of the Netherlands
A holder of ordinary shares will not be subject to Netherlands taxes on income or on capital gains in respect of any payment under the ordinary shares or any gain realized on the disposal or deemed disposal of the ordinary shares, provided that:
| (a) | such holder is neither a resident nor deemed to be resident in the Netherlands for Dutch tax purposes and, if such holder is an individual, such holder does not qualify for the application of the rules of the Dutch Income Tax Act 2001 as they apply to residents of the Netherlands; |
| (b) | such holder does not have an interest in an enterprise or a deemed enterprise (statutorily defined term) which, in whole or in part, is either effectively managed in the Netherlands or is carried out through a permanent establishment, a deemed permanent establishment or a permanent representative in the Netherlands and to which enterprise or part of an enterprise the ordinary shares are attributable or deemed attributable or has a deemed enterprise for activities performed as statutory director (‘bestuurder’) or supervisory director (‘commissaris’) of a company resident of the Netherlands; and |
| (c) | in the event such holder is an individual, such holder does not carry out any activities in the Netherlands with respect to the ordinary shares that go beyond ordinary asset management and does not derive benefits from the ordinary shares that are taxable as benefits from other activities in the Netherlands. |
(c) Gift and Inheritance Taxes
(i) Residents of the Netherlands
Gift and inheritance taxes will arise in the Netherlands with respect to a transfer of the ordinary shares by way of a gift by, or on the death of, a holder of ordinary shares who is resident or deemed to be resident in the Netherlands at the time of the gift or his/her death.
(ii) Non-residents of the Netherlands
No Dutch gift or inheritance taxes will arise on the transfer of the ordinary shares by way of gift by, or on the death of, a holder of ordinary shares who is neither resident nor deemed to be resident in the Netherlands, unless:
| (a) | in the case of a gift of ordinary shares by an individual who at the date of the gift was neither resident nor deemed to be resident in the Netherlands, such individual dies within 180 days after the date of the gift, while being resident or deemed to be resident in the Netherlands; or |
| (b) | the transfer is otherwise construed as a gift or inheritance made by, or on behalf of, a person who, at the time of the gift or death, is or is deemed to be resident in the Netherlands. |
For purposes of Dutch gift and inheritance taxes, amongst others, a person that holds the Dutch nationality will be deemed to be resident in the Netherlands if such person has been resident in the Netherlands at any time during the ten years preceding the date of the gift or his/her death. Additionally, for purposes of Dutch gift tax, amongst others, a person not holding the Dutch nationality will be deemed to be resident in the Netherlands if such person has been resident in the Netherlands at any time during the twelve months preceding the date of the gift. Applicable tax treaties may override deemed residency.
(d) Other Taxes and Duties
No Dutch VAT and no Dutch registration tax, stamp duty or any other similar documentary tax or duty will be payable by a holder of ordinary shares on any payment in consideration for the holding or disposal of the ordinary shares.
129
(e) Residence
A shareholder will not become resident or deemed resident in the Netherlands for tax purposes by reason only of holding the ordinary shares.
Certain Material U.S. Federal Income Tax Considerations
The following is a summary of certain material U.S. federal income tax considerations relating to the ownership and disposition of our ordinary shares by a U.S. holder (as defined below). This summary addresses only the U.S. federal income tax considerations for U.S. holders that will hold our ordinary shares as capital assets. This summary does not address all U.S. federal income tax matters that may be relevant to a particular U.S. holder. This summary does not address tax considerations applicable to a holder of ordinary shares that may be subject to special tax rules including, without limitation, the following:
| ● | banks, financial institutions or insurance companies; |
| ● | brokers, dealers or traders in securities, currencies, commodities, or notional principal contracts; |
| ● | tax-exempt entities or organizations, including an “individual retirement account” or “Roth IRA” as defined in Section 408 or 408A of the Code (as defined below), respectively; |
| ● | real estate investment trusts, regulated investment companies, or grantor trusts; |
| ● | persons that hold the ordinary shares as part of a “hedging,” “integrated”, or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes; |
| ● | partnerships (including entities classified as partnerships for U.S. federal income tax purposes) or other pass-through entities (including S corporations), or persons that will hold our shares through such an entity; |
| ● | certain former citizens or long-term residents of the United States; |
| ● | persons that received our shares as compensation for the performance of services; |
| ● | persons that acquire ordinary shares as a result of holding or owning our preferred shares; |
| ● | holders that own directly, indirectly, or through attribution 10% or more of the voting power or value of our shares; and |
| ● | holders that have a “functional currency” other than the U.S. dollar. |
Further, this summary does not address the U.S. federal estate, gift, or alternative minimum tax considerations, or any U.S. state, local, or non-U.S. tax considerations (other than the Dutch tax considerations discussed above) of the ownership and disposition of our ordinary shares.
This description is based on the U.S. Internal Revenue Code of 1986, as amended, or the Code, existing, proposed, and temporary U.S. Treasury Regulations promulgated thereunder, and administrative and judicial interpretations thereof, in each case as in effect on the date hereof. All the foregoing is subject to change, which change could apply retroactively, and to differing interpretations, all of which could affect the tax considerations described below. There can be no assurances that the U.S. Internal Revenue Service, or the IRS, will not take a contrary position concerning the tax consequences of the ownership and disposition of our ordinary shares or that such a position would not be sustained. Holders should consult their own tax advisers concerning the U.S. federal, state, local, and non-U.S. tax consequences of owning and disposing of our ordinary shares in their particular circumstances.
130
For purposes of this summary, a “U.S. holder” is a beneficial owner of ordinary shares that is (or is treated as), for U.S. federal income tax purposes:
| ● | a citizen or resident of the United States; |
| ● | a corporation, or other entity that is treated as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia; |
| ● | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| ● | a trust, if a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of the substantial decisions of such trust, or if the trust has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a United States person. |
If a partnership (or any other entity treated as a partnership for U.S. federal income tax purposes) holds ordinary shares, the U.S. federal income tax consequences relating to an investment in our ordinary shares will depend in part upon the status of the partner and the activities of the partnership. Such a partner or partnership should consult its tax advisor regarding the U.S. federal income tax considerations of owning and disposing of our ordinary shares in its particular circumstances.
As indicated below, this discussion is subject to U.S. federal income tax rules applicable to a “passive foreign investment company,” or a PFIC.
Persons considering an investment in our ordinary shares should consult their own tax advisors as to the particular tax consequences applicable to them relating to the ownership and disposition of our ordinary shares, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
(a) Distributions
Subject to the discussion under “Passive Foreign Investment Company Considerations,” below, the gross amount of any distribution (including any amounts withheld in respect of Dutch withholding tax) actually or constructively received by a U.S. holder with respect to an ordinary share will be taxable to the U.S. holder as a dividend to the extent the distribution is made out of our current and accumulated earnings and profits as determined under U.S. federal income tax principles. Distributions in excess of earnings and profits will be non-taxable to the U.S. holder to the extent of, and will be applied against and reduce, the U.S. holder’s adjusted tax basis in each such ordinary share. Distributions in excess of earnings and profits and such adjusted tax basis will generally be taxable to the U.S. holder as either long-term or short-term capital gain, depending upon whether the U.S. holder has held our ordinary shares for more than one year as of the time such distribution is received. However, since we do not calculate our earnings and profits under U.S. federal income tax principles, it is expected that all distributions will be reported as dividends, even if a distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above.
Non-corporate U.S. holders may qualify for the preferential rates of taxation with respect to dividends on ordinary shares applicable to long-term capital gains (i.e., gains from the sale of capital assets held for more than one year) applicable to qualified dividend income (as discussed below). The Company, which is incorporated under the laws of the Kingdom of the Netherlands, believes that it qualifies as a resident of the Netherlands for purposes of, and is eligible for the benefits of, the Convention between the United States of America and the Kingdom of the Netherlands for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with respect to Taxes on Income, signed on December 18, 1992, as amended and currently in force, or the U.S.-Netherlands Tax Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-Netherlands Tax Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange-of-information program. Therefore, subject to the discussion under “Passive Foreign Investment Company Considerations,” below, if the U.S.-Netherlands Tax Treaty is applicable, such dividends will generally be “qualified dividend income” in the hands of individual U.S. holders, provided that certain conditions are met, including the holding period requirement as well as the absence of certain risk reduction transactions.
131
A U.S. holder generally may claim the amount of Dutch income withholding tax withheld as either a deduction from gross income or a credit against U.S. federal income tax liability. However, the foreign tax credit is subject to numerous complex limitations that must be determined and applied on a case-by-case basis. Each U.S. holder should consult its own tax advisors regarding the foreign tax credit rules.
In general, the amount of a distribution paid to a U.S. holder in a foreign currency will be the dollar value of the foreign currency calculated by reference to the spot exchange rate on the day the U.S. holder receives the distribution, regardless of whether the foreign currency is converted into U.S. dollars at that time. Any foreign currency gain or loss a U.S. holder realizes on a subsequent conversion of foreign currency into U.S. dollars will be U.S. source ordinary income or loss. If dividends received in a foreign currency are converted into U.S. dollars on the day they are received, a U.S. holder should not be required to recognize foreign currency gain or loss in respect of the dividend.
(b) Sale, exchange or other taxable disposition of our ordinary shares
A U.S. holder will generally recognize gain or loss for U.S. federal income tax purposes upon the sale, exchange, or other taxable disposition of an ordinary share in an amount equal to the difference between the U.S. dollar value of the amount realized from such sale or exchange and the U.S. holder’s tax basis in that ordinary share. Subject to the discussion under “Passive Foreign Investment Company Considerations” below, this gain or loss will generally be a capital gain or loss and will generally be treated as from sources within the United States. The adjusted tax basis in an ordinary share generally will equal the cost of such ordinary share. Capital gain from the sale, exchange, or other taxable disposition of ordinary shares by a non-corporate U.S. holder is generally eligible for a preferential rate of taxation applicable to capital gains, if the non-corporate U.S. holder’s holding period determined at the time of such sale, exchange, or other taxable disposition for such ordinary share exceeds one year (i.e., such gain is long-term taxable gain). The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations under the Code. Any such gain or loss that a U.S. holder recognizes generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes.
For a cash basis taxpayer, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the settlement date of the purchase or sale. In that case, no foreign currency exchange gain or loss will result from currency fluctuations between the trade date and the settlement date of such a purchase or sale. An accrual basis taxpayer, however, may elect the same treatment required of cash basis taxpayers with respect to purchases and sales of our ordinary shares that are traded on an established securities market, provided the election is applied consistently from year to year. Such election may not be changed without the consent of the IRS. For an accrual basis taxpayer that does not make such an election, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the trade date of the purchase or sale. Such an accrual basis taxpayer may recognize exchange gain or loss based on currency fluctuations between the trade date and the settlement date. Any foreign currency gain or loss a U.S. holder realizes will be U.S. source ordinary income or loss.
(c) Net Investment Income Tax
Certain U.S. holders that are individuals, estates or trusts are subject to a 3.8% tax on all or a portion of their “net investment income,” which may include all or a portion of their dividend income and net gains from the disposition of ordinary shares. Each U.S. holder that is an individual, estate or trust is urged to consult its tax advisors regarding the applicability of the Net Investment Income Tax to its income and gains in respect of its investment in our ordinary shares.
(d) Passive foreign investment company considerations
If we are classified as a passive foreign investment company, or PFIC, in any taxable year, a U.S. holder would be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. holder could derive from investing in a non-U.S. company that does not distribute all its earnings on a current basis. We will inform our shareholders in our annual report on Form 20-F if we determine that we are a PFIC.
132
A corporation organized outside the United States generally will be classified as a PFIC for U.S. federal income tax purposes in any taxable year in which either: (i) at least 75% of its gross income is “passive income,” referred to herein as the Income Test or (ii) at least 50% of the average quarterly value of its total gross assets (for which purpose, assuming we are treated as a publicly traded company pursuant to Section 1297(e)(3) of the Code, the total value of our assets may be determined in part by the market value of our ordinary shares, which is subject to change) is attributable to assets that produce “passive income” or are held for the production of “passive income,” Referred to herein as the Asset Test.
Passive income for this purpose generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income, and amounts derived by reason of the temporary investment of funds raised in offerings of our ordinary shares. For purposes of the PFIC tests, gross income and gross assets of a corporation include its proportionate share of the gross income and gross assets of any other corporation of which it owns directly or indirectly at least 25% by value of the stock.
If we are classified as a PFIC in any year with respect to which a U.S. holder owns our ordinary shares, we will continue to be treated as a PFIC with respect to such U.S. holder in all succeeding years during which the U.S. holder owns our ordinary shares, regardless of whether we continue to meet the tests described above, unless the U.S. holder makes a purging election, which allows a shareholder to purge the continuing PFIC taint by either making a deemed sale election or, under certain conditions, a deemed dividend election. In addition, if we are classified as a PFIC, then a U.S. holder of our shares will be deemed to own, proportionately, shares we own of lower-tier corporations. If any such lower-tier corporation is a PFIC, then a U.S. holder will be treated as an (indirect) shareholder of that lower-tier PFIC.
Based on the average value of our gross assets and the composition of our income, we believe that we were a PFIC during the 2023 taxable year. Our status for any taxable year will depend on our assets and activities in each year, and because this is a factual determination made annually after the end of each taxable year, there can be no assurance on whether we will be considered a PFIC for the current taxable year or any future taxable year. The market value of our assets may be determined in large part by reference to the market price of our ordinary shares, which is likely to fluctuate.
If we are a PFIC, and you are a U.S. holder, then unless you make one of the elections described below, a special tax regime will apply to any “excess distribution” by us to you (generally, your portion of distributions in any year which are greater than 125% of the average annual distribution received by you in the shorter of the three preceding years or your holding period for our ordinary shares). In determining the average annual distribution, the portion of any excess distribution from a prior year that was allocated to the prior-year PFIC period is disregarded. That special regime will also apply to any gain realized on the sale or other disposition of the ordinary shares. Under this special regime, any excess distribution and realized gain will be treated as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period, (b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for such year applicable to you (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to tax at the U.S. holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed below), and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to long-term capital gains discussed above under “Distributions.”
133
Certain elections exist that may alleviate some of the adverse consequences of PFIC status and would result in an alternative treatment (such as mark-to-market treatment) of the common shares. If a U.S. holder makes the mark-to-market election, the U.S. holder generally will recognize as ordinary income any excess of the fair market value of the ordinary shares at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ordinary shares over their fair market value at the end of the taxable year, but only to the extent of the net amount of income previously included as a result of the mark-to-market election and not offset by prior mark‐to market losses. If a U.S. holder makes the election, the U.S. holder’s tax basis in the ordinary shares will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale or other disposition of ordinary shares in a year when we are a PFIC will be treated as ordinary income, and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). The mark-to-market election is available only if we are a PFIC and our ordinary shares are “regularly traded” on a “qualified exchange.” Our ordinary shares will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ordinary shares are traded on a qualified exchange on at least 15 days during each calendar quarter (subject to the rule that trades that have as one of their principle purposes the meeting of the trading requirement as disregarded). The Nasdaq Capital Market is a qualified exchange for this purpose and, consequently, if the ordinary shares are regularly traded on that market, the mark-to-market election will be available to a U.S. holder. U.S. holders should consult their tax advisors to determine whether the mark-to-market election would be available and, if so, what the consequences of making that election would be in their particular circumstances.
Alternatively, you may avoid the general tax treatment for PFICs described above by electing to treat us as a “qualified electing fund” under Section 1295 of the Code, or QEF, for each of the taxable years during your holding period that we are a PFIC. If a QEF election is not in effect for the first taxable year in your holding period in which we are a PFIC, a QEF election generally can only be made if you elect to make an applicable deemed sale or deemed dividend election on the first day of your taxable year in which the PFIC becomes a QEF pursuant to the QEF election. The deemed gain or deemed dividend recognized with respect to such an election would be subject to the general tax treatment of PFICs discussed above. We intend to determine our PFIC status at the end of each taxable year and to satisfy any applicable record keeping and reporting requirements that apply to a QEF, and will endeavor to provide to you, for each taxable year that we determine we are a PFIC, a PFIC Annual Information Statement containing information necessary for you to make a QEF election with respect to us. We may elect to provide such information on our website.
If you make a QEF election with respect to a PFIC, you will be taxed currently on your pro rata share of the PFIC’s ordinary earnings and net capital gain (at ordinary income and capital gain rates, respectively) for each taxable year that the entity is a PFIC, even if no distributions are received. Any distributions we make out of our earnings and profits that were previously included in your income under the QEF election would not be taxable to you. Your tax basis in your common shares would be increased by an amount equal to any income included under the QEF election and decreased by any amount distributed on the common shares that is not included in your income. In addition, you will recognize capital gain or loss on the disposition of your common shares in an amount equal to the difference between the amount realized and your adjusted tax basis in the common shares, each as determined in U.S. dollars. Once made, a QEF election remains in effect unless invalidated or terminated by the IRS or revoked by the shareholder. A QEF election can be revoked only with the consent of the IRS. You will not be currently taxed on the ordinary income and net capital gain of a PFIC with respect to which a QEF election was made for any taxable year of the non-U.S. corporation that such corporation does not satisfy the PFIC Income Test or Asset Test. You are urged to consult your own tax advisors regarding the availability of, and procedure for making, any deemed gain, deemed dividend or QEF election.
If we are a PFIC, the general tax treatment for U.S. holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. holders who are indirect shareholders of lower-tier PFICs, as discussed above.
If a U.S. holder owns ordinary shares during any taxable year in which we are a PFIC, the U.S. holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) with respect to the company (and with respect to any lower-tier PFICs) with the U.S. holder’s federal income tax return for that year. If our company were a PFIC for a given taxable year, then you should consult your tax advisor concerning your annual filing requirements.
134
The U.S. federal income tax rules relating to PFICs are complex. U.S. investors are urged to consult their own tax advisers with respect to the ownership and disposition of our ordinary shares, the consequences to them of an investment in a PFIC, any elections available with respect to our ordinary shares, and the IRS information reporting obligations with respect to the ownership and disposition of our ordinary share.
(e) Backup Withholding and Information Reporting
U.S. holders generally will be subject to information reporting requirements with respect to dividends on ordinary shares and on the proceeds from the sale, exchange, or disposition of ordinary shares that are paid within the United States or through U.S.-related financial intermediaries, unless the U.S. holder is an “exempt recipient.” In addition, U.S. holders may be subject to backup withholding on such payments, unless the U.S. holder provides a taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an exemption. Backup withholding is not an additional tax, and the amount of any backup withholding will be allowed as a credit against a U.S. holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
(f) Foreign Asset Reporting
Certain U.S. holders who are individuals are required to report information relating to an interest in our ordinary shares, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions) by filing IRS Form 8938 (“Statement of Specified Foreign Financial Assets”) with their federal income tax return. U.S. holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of our ordinary shares.
THE DISCUSSION SET OUT ABOVE IS INCLUDED FOR GENERAL INFORMATION ONLY AND MAY NOT BE APPLICABLE DEPENDING UPON A HOLDER’S PARTICULAR SITUATION. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE RELEVANT TO A HOLDER. EACH HOLDER IS URGED TO CONSULT THEIR TAX ADVISOR WITH RESPECT TO THE TAX CONSEQUENCES OF THE OWNERSHIP AND DISPOSITION OF THE SHARES INCLUDING TAX CONSEQUENCES UNDER STATE, LOCAL AND OTHER TAX LAWS AND THE POSSIBLE TAX EFFECTS OF CHANGES IN THE UNITED STATES FEDERAL AND OTHER TAX LAWS.
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
H. Documents on Display
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge on the websites described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F containing financial statements that have been examined and reported on, with an opinion expressed by an independent registered public accounting firm.
135
We maintain a corporate website at www.ProQR.com. We intend to post our Annual Report on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report. We have included our website address in this Annual Report solely as an inactive textual reference.
The SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as ProQR, that file electronically with the SEC.
With respect to references made in this Annual Report to any contract or other document relating to ProQR, such references are not necessarily complete and you should refer to the exhibits attached or incorporated by reference to this Annual Report for copies of the actual contract or document
I. Subsidiary information
Not applicable.
J. Annual Report to Security Holders
Not applicable.
Item 11: Quantitative and Qualitative Disclosures about Market Risk
We are exposed to a variety of financial risks: market risk (including foreign exchange risk and interest rate risk), credit risk and liquidity risk. Our overall risk management program focuses on the unpredictability of financial markets.
Our cash management policy is focused on optimizing the net interest result of funds invested in deposits while accepting limited risk (minimum ratings of P-1 or A2 for short-term and long-term, respectively by Moody’s and A-1 or A for short-term and long-term, respectively, by Standard and Poor’s). Individual commitments exceeding a defined threshold in foreign currencies may be hedged against our functional currency, the euro, to avoid foreign currency exposure affecting the income statement in comparison to budget.
Market Risk
Foreign exchange risk arises from future commercial transactions and recognized assets and liabilities in foreign currencies, primarily with respect to the U.S. Dollar. We currently do not engage in hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the euro. We have exposure associated with the time delay between entering into a contract, budget or forecast and the realization thereof. To minimize economic exposure related to currency fluctuations, currencies on the balance sheet will be matched to the cash flows on an annual basis to ensure the availability of at least 12 months of liquidity per currency per the approved budget. The impact of fair value measurements on the financial statements is limited.
At December 31, 2023 our net position of financial instruments denominated in U.S. Dollars was a net liability of € 726,000 (2022: net asset of € 78,630,000). Foreign currency denominated receivables and trade payables are short term in nature (generally 30 to 45 days). As a result foreign exchange rate movements on receivables and trade payables, during the years presented had an immaterial effect on the financial statements.
Our interest rate risk exposure is limited due to the use of loans with fixed rates. At December 31, 2023, we had a loan with a fixed interest rate, totaling € 4,292,000 (2022: several loans totaling € 6,771,000).
Credit Risk
Credit risk represents the risk of financial loss caused by default of the counterparty. We have no large receivables balances with external parties, except with Eli Lilly and Company. Our principal financial assets are cash and cash equivalents which are, in line with our cash policy, placed at banks which meet our defined minimum credit ratings.
136
Liquidity Risk
Management forecasts our liquidity requirements to ensure that there is sufficient cash to meet operational needs. We believe that our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements into mid-2026. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect.
Item 12: Description of Securities other than Equity Securities
A. Debt Securities
Not applicable.
B. Warrants and Rights
Not applicable.
C. Other Securities
Not applicable.
D. American Depositary Shares
Not applicable.
137
Part II
Item 13: Defaults, Dividend Arrearages and Delinquencies
No matters to report.
Item 14: Material Modifications to the Rights of Security Holders and Use of Proceeds
No matters to report.
Item 15: Controls and Procedures
A. Disclosure Controls and Procedures
Under the supervision and with the participation of our Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”), the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934) has been evaluated as of the end of the period covered by the Annual Report (December 31, 2023). The term “disclosure controls and procedures” means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms and that such information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosures.
Based on that evaluation, our CEO and CFO have concluded that these disclosure controls and procedures are effective as at December 31, 2023.
B. Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate "internal control over financial reporting," as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. This rule defines internal control over financial reporting as a process designed by, or under the supervision of, a company’s chief executive officer and chief financial officer and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
The Company’s internal control over financial reporting is a process designed to provide reasonable, but not absolute, assurance regarding the reliability of financial reporting and the preparation of the consolidated financial statements in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting is not intended to provide absolute assurance that a misstatement of our financial statements would be prevented or detected.
In connection with the preparation of the Company’s annual consolidated financial statements, management has undertaken an assessment of the effectiveness of the Company’s internal control over financial reporting as of December 31, 2023. Based on this assessment, management concluded that the Company’s internal control over financial reporting was effective as at December 31, 2023.
138
C. Attestation report of the registered public accounting firm
This Annual Report includes an attestation report of the company’s registered public accounting firm on the effectiveness of the Company’s internal control over financial reporting.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Supervisory Board
ProQR Therapeutics N.V.:
Opinion on Internal Control Over Financial Reporting
We have audited ProQR Therapeutics N.V. and subsidiaries’ (the “Company”) internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated statements of financial position of the Company as of December 31, 2023 and 2022, the related consolidated statements profit or loss and comprehensive income, changes in equity, and cash flows for each of the years in the three year period ended December 31, 2023, and the related notes (collectively, the consolidated financial statements), and our report dated March 13, 2024 expressed an unqualified opinion on those consolidated financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
139
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ KPMG Accountants N.V.
Amstelveen, the Netherlands
March 13, 2024
D. Changes in Internal Control over Financial Reporting
During the year ended December 31, 2023, there have been no changes in our internal control over financial reporting that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 16: [Reserved]
Item 16A: Audit Committee Financial Expert
Currently, Bart Filius qualifies as an “audit committee financial expert,” as defined by the SEC and as determined by our supervisory board. In addition, he satisfies the independence requirements of the Nasdaq listing standards and Rule 10A-3(b)(1) under the Exchange Act and the criteria for independence set forth in best practice 2.1.8 of the DCGC.
Item 16B: Code of Ethics
We have adopted a Code of Business Conduct and Ethics applicable to all of our management board and supervisory board members and our employees, including our CEO, CFO, controller or principal accounting officers, or other persons performing similar functions, which is a “code of ethics” as defined by the SEC. The full text of the Code of Business Conduct and Ethics is posted on our company website at www.ProQR.com.
If we make any amendment to the Code of Business Conduct and Ethics or grant any waivers, including any implicit waiver, from a provision of the Code of Business Conduct and Ethics for our Supervisory Board members, Management Board members or other executive officers, we will disclose the nature of such amendment or waiver on our website within five working days to the extent required by the rules and regulations of the SEC.
The Code of Business Conduct and Ethics includes the whistleblower policy as contemplated in the DCGC.
Item 16C: Principal Accountant Fees and Services
For the years ended December 31, 2023, 2022 and 2021, our independent public accounting firm is
The information required is included in Note 28 to the financial statements included elsewhere in this Annual Report.
Item 16D: Exemptions from the Listing Standards for Audit Committees
Not applicable.
Item 16E: Purchases of Equity Securities by the Issuer and Affiliated Purchasers
In 2023, no purchases of our registered equity securities were made by us or on our behalf or on the behalf of any affiliated purchaser.
140
Item 16F: Change in Registrant’s Certifying Accountant
Not applicable.
Item 16G: Corporate Governance
The Sarbanes-Oxley Act of 2002, as well as related rules subsequently implemented by the SEC, requires foreign private issuers, including our company, to comply with various corporate governance practices. As a foreign private issuer, subject to certain exceptions, the Nasdaq listing standards permit a foreign private issuer to follow its home country practice in lieu of the Nasdaq listing standards. In addition, a foreign private issuer must disclose in its annual reports filed with the SEC each such requirement that it does not follow and describe the home country practice followed instead of any such requirement.
The home country practices followed by our company in lieu of Nasdaq rules are described below:
| ● | We do not intend to follow Nasdaq’s quorum requirements applicable to meetings of shareholders. In accordance with Dutch law and generally accepted business practice, our articles of association do not provide quorum requirements generally applicable to general meetings of shareholders. |
| ● | We do not intend to follow Nasdaq’s requirements regarding the provision of proxy statements for general meetings of shareholders. Dutch law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in the Netherlands. We do intend to provide shareholders with an agenda and other relevant documents for the general meeting of shareholders and shareholders will be entitled to give proxies and voting instructions to us and/or third parties. |
| ● | We do not intend to follow Nasdaq’s requirements regarding the independence of all members of the compensation, nominating and corporate governance committee. Dutch law and the DCGC do not require that the compensation, nominating and corporate governance committee be composed entirely of independent directors and only requires a mere majority. In accordance with Dutch law and the DCGC, our compensation, nominating and corporate governance committee consists of a majority of members who qualify as independent under applicable Nasdaq standards and under the DCGC. |
| ● | We do not intend to follow Nasdaq’s requirements regarding Nasdaq Listing Rule 5605(b)(2), which mandates that independent directors must meet at regularly scheduled executive sessions where only independent directors are present. In accordance with Dutch law and the DCGC, our directors may choose to meet in executive sessions at their discretion. |
We intend to take all actions necessary for us to maintain compliance as a foreign private issuer under the applicable corporate governance requirements of the Sarbanes-Oxley Act of 2002, the rules adopted by the SEC and Nasdaq’s listing standards.
Certain Nasdaq corporate governance requirements are not reflected in the DCGC or Dutch law. Accordingly, our shareholders may not be afforded the same protection as provided under Nasdaq corporate governance rules to the extent the DCGC or Dutch law does not provide similar protections. Furthermore, as a foreign private issuer, we will be exempt from the rules and regulations under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders will be exempt from the reporting and short swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we will not be required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as domestic companies whose securities are registered under the Exchange Act.
Item 16H: Mine Safety Disclosure
Not applicable.
141
Item 16I: Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
Item 16J: Insider trading policies
Not applicable.
Item 16K: Cybersecurity
ProQR uses, stores and processes data for and about our employees, partners and suppliers. We have implemented an information security management framework (“ISM”) – which includes policies, operating procedures, and processes – that is designed to identify, assess, and mitigate risks from cybersecurity threats to this data and our systems.
Governance Related to Cybersecurity Risks
Our management board has primary responsibility for implementing our cybersecurity risk management program, as set forth in the ISM. Our management board has tasked the Chief People and Operations Officer (“CPOO”) as the member of the executive committee with day-to-day management of the ISM. Assisting the CPOO in managing the ISM is an ISM Committee of senior team members and advisors, which currently consists of the CPOO and representatives from information technology, legal, and privacy.
Our IT Director has responsibility for overseeing and implementing the cybersecurity program and reports directly to the CPOO, and has over 20 years of experience in the field of IT, including in network and systems security. We have also appointed an Information Security Officer to assist in managing our cybersecurity threat management program. Our Information Security Officer has broad experience in the IT field for over 30 years, including with respect to information security, internal audits, training and SOX compliance. Additionally, our Information Security Officer has a range of IT- and cybersecurity-related certifications issued by the Professional Evaluation and Certification Board.
Our audit committee, per the stipulations of our audit committee charter, oversees the ISM, including the assessment of data governance, security initiatives, significant existing and emerging cybersecurity risks, cybersecurity incidents, and any disclosure obligations arising from such incidents, as applicable.
Our audit committee convenes periodically to assess the company’s applications of information and communication technology, including risks relating to cybersecurity, as appropriate. The CPOO and ISM Committee report to our audit committee on relevant cybersecurity matters and associated risks as needed.
Cyber Risk Management and Strategy
Under the guidance of the ISM Committee and the Information Security Officer, and with the support of our third-party information technology providers, our ISM addresses areas such as:
| ● | Assessment and treatment of cyber and information security risks |
| ● | Assessment and treatment of information technology service continuity risks |
| ● | Assessment of cybersecurity risks related to certain third-party vendors, pursuant to our Vendor Management Policy |
Additionally, we have an employee education program that is designed to raise awareness of cybersecurity threats to reduce our vulnerability to such threats as well as to encourage consideration of cybersecurity risks across functions. Further, we maintain an Incident Management Policy designed to assist us in identifying, responding to, and recovering from cybersecurity incidents.
142
As of the date of this report, we have not identified cybersecurity threats that have materially affected or are reasonably likely to materially affect the Company, including our business strategy, results of operations, or financial condition. However, we have from time to time, experienced threats and security incidents relating to our and our third-party vendors’ information systems. For information regarding cybersecurity risks that may affect the Company, see Part I, Item 3.D: “Risk Factors— Risks Related to Our Organization, Structure and Operations” in this Annual Report.
Part III
Item 17: Financial Statements
Financial Statements are filed as part of this Annual Report, starting on page F-1.
Item 18: Financial Statements
Financial Statements are filed as part of this Annual Report, starting on page F-1.
Item 19: Exhibits
The Exhibits listed in the Exhibit Index at the end of this Annual Report are filed as Exhibits to this Annual Report.
143
Index of Exhibits
Exhibit No. |
| Description |
|---|---|---|
1.1 | ||
1.2 | ||
1.3 | ||
2.1 | ||
2.2 | ||
2.3 | ||
4.1# | ||
4.2# | ||
4.3# | ||
4.4 | ||
4.5# | ||
4.6 |
144
Exhibit No. |
| Description |
|---|---|---|
4.7†† | ||
4.8† | ||
4.9† | ||
4.10† | ||
4.11† | ||
4.12†† | ||
4.13†† | ||
4.14†† | ||
4.15 | ||
4.16 | ||
4.17† | ||
145
Exhibit No. |
| Description |
|---|---|---|
4.18 | ||
4.19 | ||
4.20 | ||
4.21 | ||
4.22 | ||
8.1* | ||
12.1* | ||
12.2* | ||
13.1** | ||
15.1* | Consent of KPMG Accountants N.V., Independent Registered Public Accounting Firm | |
97.1* | ||
101.INS* | XBRL Instance Document. | |
101.SCH* | XBRL Taxonomy Extension Schema Document. | |
101.CAL* | XBRL Taxonomy Extension Calculation Linkbase Document. | |
101.DEF* | XBRL Taxonomy Extension Definition Linkbase Document. | |
101.LAB* | XBRL Taxonomy Extension Label Linkbase Document. | |
101.PRE* | XBRL Taxonomy Extension Presentation Linkbase Document. | |
104* | Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
146
* | Filed herewith |
** | Indicates that the exhibit is being furnished and will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to the liability of that section. Such exhibit will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933, as amended, or the Exchange Act, except to the extent specifically incorporated by reference into such filing. |
† | Confidential treatment has been granted as to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission. |
†† | Certain confidential portions of this exhibit (indicated by brackets and asterisks) have been omitted from this exhibit. |
# | Indicates management contract or compensatory plan or arrangement. |
147
Signatures
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
Leiden, March 13, 2024
PROQR THERAPEUTICS N.V. | ||
By: | /s/ Daniel de Boer | |
Name: | Daniel de Boer | |
Title: | Chief Executive Officer | |
By: | /s/ Jurriaan Dekkers | |
Name: | Jurriaan Dekkers | |
Title: | Chief Financial Officer | |
148
INDEX TO FINANCIAL STATEMENTS
PAGE | |
Consolidated Financial Statements as at December 31, 2023 and 2022 and for the Years Ended December 31, 2023, December 31, 2022 and December 31, 2021 |
|
F-2 | |
F-3 | |
Consolidated Statement of Profit or Loss and Comprehensive Income | F-4 |
F-5 | |
F-6 | |
F-7 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the supervisory board
ProQR Therapeutics N.V:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statements of financial position of ProQR Therapeutics N.V. and subsidiaries (the “Company”) as of December 31, 2023 and 2022, the related consolidated statements of profit or loss and comprehensive income, changes in equity, and cash flows for each of the years in the three-year period ended December 31, 2023, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2023, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company’s internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 13, 2024 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/ KPMG Accountants N.V.
We have served as the Company's auditor since 2020.
Amstelveen, The Netherlands
March 13, 2024
F-2
PROQR THERAPEUTICS N.V.
Consolidated Statement of Financial Position
|
| December 31, |
| December 31, | ||
2023 | 2022 | |||||
(€ in thousands) | ||||||
Assets |
|
|
|
|
|
|
Property, plant and equipment |
| 7 |
| |
| |
Investments in financial assets | 9 | — | | |||
Non-current assets |
|
|
| |
| |
Other taxes |
| 10 |
| |
| |
Prepayments and other receivables |
| 11 |
| | | |
Cash and cash equivalents |
| 12 |
| |
| |
Current assets |
|
|
| |
| |
Total assets |
|
|
| |
| |
Shareholders’ equity |
|
|
|
|
|
|
Share capital |
|
|
| |
| |
Share premium |
|
|
| |
| |
Reserves |
|
|
| |
| |
Accumulated deficit |
|
|
| ( |
| ( |
Equity attributable to owners of the Company |
| 13 |
| |
| |
Non-controlling interests | — | ( | ||||
Total equity | | | ||||
Liabilities |
|
|
|
|
|
|
Borrowings |
| 14 |
| |
| |
Lease liabilities |
| 25 |
| |
| |
Deferred income | 15 | | | |||
Non-current liabilities |
|
| |
| | |
Borrowings | 14 | — | | |||
Lease liabilities | 25 | | | |||
Derivative financial instruments | 14 | | | |||
Trade payables |
|
|
| |
| |
Social securities and other taxes |
|
| |
| | |
Deferred income |
| 15 |
| |
| |
Other current liabilities |
| 16 |
| |
| |
Current liabilities |
|
| |
| | |
Total liabilities |
|
|
| |
| |
Total equity and liabilities |
|
|
| |
| |
The accompanying notes are an integral part of these consolidated financial statements.
F-3
PROQR THERAPEUTICS N.V.
Consolidated Statement of Profit or Loss and Comprehensive Income
Year Ended December 31, | |||||||||||
|
| 2023 |
| 2022 |
| 2021 | |||||
(€ in thousands) | |||||||||||
Revenue | 17 | | | | |||||||
Other income |
| 18 |
| |
| |
| | |||
Research and development costs |
|
| ( |
| ( |
| ( | ||||
General and administrative costs |
|
|
| ( |
| ( |
| ( | |||
Total operating costs |
| 19 |
| ( |
| ( |
| ( | |||
Operating result |
|
|
| ( |
| ( |
| ( | |||
Financial income |
| 21 |
|
| |
| |
| | ||
Financial expense | 21 | ( | ( | ( | |||||||
Results related to associates | 8 | — | ( | ( | |||||||
Gain on disposal of associate | 9 | — | — | | |||||||
Gain on disposal of subsidiary | 14 | | — | — | |||||||
Results related to derecognition of financial liabilities | 14 | | ( | — | |||||||
Results related to financial liabilities measured at FVTPL | 22 | | | ( | |||||||
Result before corporate income taxes |
|
|
| ( |
| ( |
| ( | |||
Corporate income taxes |
| 23 |
| |
| ( |
| ( | |||
Result for the year |
|
|
| ( |
| ( |
| ( | |||
Other comprehensive income |
|
|
|
|
|
|
| ||||
Items that will never be reclassified to profit or loss |
|
|
| ||||||||
Fair value loss on investment in financial asset designated as at FVTOCI | ( | — | — | ||||||||
Items that are or may be reclassified to profit or loss |
|
|
|
|
|
| |||||
Foreign operations – foreign currency translation differences |
|
|
| ( |
| |
| | |||
Total comprehensive loss |
|
|
| ( |
| ( |
| ( | |||
|
| ||||||||||
Result attributable to |
|
| |||||||||
Owners of the Company | ( |
| ( |
| ( | ||||||
Non-controlling interests | |
| |
| ( | ||||||
( |
| ( |
| ( | |||||||
|
| ||||||||||
Total comprehensive loss attributable to |
|
| |||||||||
Owners of the Company | ( |
| ( |
| ( | ||||||
Non-controlling interests | |
| |
| ( | ||||||
( |
| ( |
| ( | |||||||
Share information |
| 24 |
|
|
|
|
| ||||
Weighted average number of shares outstanding |
|
|
| |
| |
| | |||
Earnings per share for result attributable to the equity holders of the Company (expressed in Euro per share) |
|
|
|
|
|
|
| ||||
Basic and diluted loss per share |
|
| € | ( | € | ( | ( | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
PROQR THERAPEUTICS N.V.
Consolidated Statement of Changes in Equity
Attributable to Equity Holders of the Company | ||||||||||||||||||
Equity Settled | Option | |||||||||||||||||
Employee | premium on | |||||||||||||||||
Share | Share | Benefit | convertible | Translation | Accumulated | Non-controlling | Total | |||||||||||
Capital | Premium | Reserve | loans | Reserve | Deficit | Total | Interests | Equity | ||||||||||
| (€ in thousands) |
| (€ in thousands) |
| (€ in thousands) |
| (€ in thousands) | (€ in thousands) | (€ in thousands) |
| (€ in thousands) | (€ in thousands) | (€ in thousands) | |||||
Balance at January 1, 2021 |
| |
| |
| |
| | ( | ( |
| | ( | | ||||
Result for the year |
| — | — | — | — | — | ( |
| ( | ( | ( | |||||||
Other comprehensive income |
| — | — | — | — | | — |
| | — | | |||||||
Recognition of share-based payments |
| | | | — | — | — |
| | — | | |||||||
Issue of ordinary shares | | | — | — | — | — | | — | | |||||||||
Equity component of convertible loan | — | — | — | | — | — | | — | | |||||||||
Shares options lapsed | — | — | ( | — | — | | — | — | — | |||||||||
Shares options exercised |
| | | ( | — | — | |
| | — | | |||||||
Balance at December 31, 2021 |
| |
| |
| |
| | | ( |
| | ( | | ||||
Result for the year |
| — | — | — | — | — | ( |
| ( | | ( | |||||||
Other comprehensive income |
| — | — | — | — | | — |
| | — | | |||||||
Recognition of share-based payments |
| — | — | | — | — | — |
| | — | | |||||||
Issue of ordinary shares | | | — | — | — | — | | — | | |||||||||
Equity component of convertible loan | — | — | — | ( | — | ( | ( | — | ( | |||||||||
Shares options lapsed | — | — | ( | — | — | | — | — | — | |||||||||
Shares options exercised |
| — | | ( | — | — | |
| | — | | |||||||
Balance at December 31, 2022 |
| |
| |
| |
| — | | ( |
| | ( | | ||||
Result for the year |
| — | — | — | — | — | ( |
| ( | | ( | |||||||
Other comprehensive loss |
| — | — | — | — | ( | ( |
| ( | — | ( | |||||||
Recognition of share-based payments |
| — | — | | — | — | — |
| | — | | |||||||
Shares options lapsed | — | — | ( | — | — | | — | — | — | |||||||||
Shares options exercised |
| — | | ( | — | — | |
| | — | | |||||||
Balance at December 31, 2023 |
| |
| |
| |
| — | | ( |
| | — | | ||||
The accompanying notes are an integral part of these consolidated financial statements. Specific reference is made to Note 13.
F-5
PROQR THERAPEUTICS N.V.
Consolidated Statement of Cash Flows
Year Ended December 31, | ||||||||
| 2023 |
| 2022 |
| 2021 | |||
(€ in thousands) | ||||||||
Cash flow from operating activities |
|
|
|
|
|
| ||
Result for the year |
| ( |
| ( |
| ( | ||
Adjustments for: |
|
|
| |||||
Other income | 18 | ( | — | — | ||||
Depreciation |
| 7 | |
| |
| | |
Results related to associates | 8 | — | | | ||||
Gain on disposal of associate | 9 | — | — | ( | ||||
Results on derecognition of subsidiary | ( | — | — | |||||
Share-based compensation |
| 13 | |
| |
| | |
Financial income and expense |
| 21 | ( |
| |
| | |
Results related to derecognition of financial liabilities | 14 | ( | | — | ||||
Results related to financial liabilities measured at fair value through profit or loss | 22 | ( | ( | | ||||
Income tax (gains) / expenses | 23 | ( | | | ||||
Changes in working capital |
| |
| ( |
| | ||
Cash generated by / (used in) operations | | ( | ( | |||||
Corporate income tax received / (paid) |
| |
| ( |
| ( | ||
Interest received |
| |
| |
| | ||
Interest paid | ( | ( | ( | |||||
Net cash generated by / (used in) operating activities |
| |
| ( |
| ( | ||
Cash flow from investing activities |
|
|
|
|
|
| ||
Purchases of property, plant and equipment |
| ( |
| ( |
| ( | ||
Proceeds from sale of property, plant and equipment | | | | |||||
Proceeds from sale of intellectual property | | — | — | |||||
Transaction costs on sale of intellectual property | ( | — | — | |||||
Net cash generated by / (used in) investing activities |
| |
| ( |
| ( | ||
Cash flow from financing activities |
|
|
|
|
|
| ||
Proceeds from issuance of shares, net of transaction costs |
| 13 | — |
| |
| | |
Proceeds from exercise of share options |
| |
| |
| | ||
Proceeds from borrowings |
| 14 | — |
| — |
| | |
Repayment of (convertible) loans | 14 | ( | ( | — | ||||
Proceeds from convertible loans | 14 | — | — | | ||||
Repayment of lease liability |
| 25 | ( |
| ( |
| ( | |
Net cash generated by / (used in) financing activities |
| ( |
| ( |
| | ||
Net increase / (decrease) in cash and cash equivalents |
| |
| ( |
| | ||
Currency effect cash and cash equivalents |
| |
| |
| | ||
Cash and cash equivalents at the beginning of the year |
| 12 | |
| |
| | |
Cash and cash equivalents at the end of the year |
| 12 | |
| |
| | |
The accompanying notes form an integral part of these consolidated financial statements.
F-6
PROQR THERAPEUTICS N.V.
Notes to the Consolidated Financial Statements
1. General Information
ProQR Therapeutics N.V. (“ProQR” or “the Company”), is a biotechnology company domiciled in the Netherlands that primarily focuses on the discovery and development of novel therapeutic medicines.
Since September 18, 2014, the Company’s ordinary shares are listed on Nasdaq. They are currently trading at Nasdaq Capital Market under ticker symbol PRQR.
The Company was incorporated in the Netherlands, on February 21, 2012 and was reorganized from a private company with limited liability to a public company with limited liability on September 23, 2014. The Company has its statutory seat in Leiden, the Netherlands and is registered in the Trade Register at the Chamber of Commerce under number 54600790. The address of its headquarters and registered office is Zernikedreef 9, 2333 CK Leiden, the Netherlands.
At December 31, 2023, ProQR Therapeutics N.V. is the ultimate parent company of the following entities:
| ● | ProQR Therapeutics Holding B.V. (the Netherlands, |
| ● | ProQR Therapeutics I B.V. (the Netherlands, |
| ● | ProQR Therapeutics II B.V. (the Netherlands, |
| ● | ProQR Therapeutics III B.V. (the Netherlands, |
| ● | ProQR Therapeutics IV B.V. (the Netherlands, |
| ● | ProQR Therapeutics V B.V. (the Netherlands, |
| ● | ProQR Therapeutics VI B.V. (the Netherlands, |
| ● | ProQR Therapeutics VII B.V. (the Netherlands, |
| ● | ProQR Therapeutics VIII B.V. (the Netherlands, |
| ● | ProQR Therapeutics IX B.V. (the Netherlands, |
| ● | ProQR Therapeutics I Inc. (United States, |
ProQR Therapeutics N.V. is also statutory director of Stichting Bewaarneming Aandelen ProQR (“ESOP Foundation”) and has full control over this entity.
As used in these consolidated financial statements, unless the context indicates otherwise, all references to “ProQR”, the “Company” or the “Group” refer to ProQR Therapeutics N.V. including its subsidiaries and the ESOP Foundation.
Revision of comparative figures
In the Company’s application of IAS 21 The Effects of Changes in Foreign Exchange Rates, certain deferred income positions were incorrectly treated as monetary items in 2021 and 2022. To correct for the effects of this error, which is immaterial for all affected prior periods, the comparative figures for the years ended December 31, 2022 and December 2021 have been revised as follows:
| ● | In the Statement of financial position as at December 31, 2022, equity attributable to owners of the Company increased by € |
| ● | In the Statement of profit or loss and other comprehensive income (“OCI”) for the years ended December 31, 2022, and December 31, 2021, revenue decreased by € |
F-7
| ● | In the Statement of changes in equity, accumulated deficit at January 1, 2022 decreased by € |
| ● | In the Statement of cash flows for the years ended December 31, 2022, and December 31, 2021, in addition to the above revisions in result for the year and net financial income and expense, changes in working capital decreased by € |
| ● | In disclosure Note 5 Financial Risk Management to the financial statements, under item (a) market risk, our net position of assets and liabilities denominated in U.S. dollars increased by € |
2. Basis of Preparation
(a) Statement of compliance
These consolidated financial statements have been prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board (“IASB”). These consolidated financial statements were authorized for issue by the Company’s Management Board and its Senior Management on March 13, 2024.
(b) Basis of measurement
The financial statements have been prepared on the historical cost basis except for financial instruments and share-based payment obligations which have been based on fair value. Historical cost is generally based on the fair value of the consideration given in exchange for assets.
(c) Functional and presentation currency
These consolidated financial statements are presented in Euro, which is the Company’s functional currency. All amounts have been rounded to the nearest thousand, unless otherwise indicated.
(d) Going concern
The management board of ProQR has, upon preparing and finalizing the 2023 financial statements, assessed the Company’s ability to fund its operations for a period of at least one year after the date of signing these financial statements. Management has not identified significant going concern risks.
The financial statements of the Company have been prepared on the basis of the going concern assumption based on its existing funding, taking into account the Company’s current cash position and the projected cash flows based on the activities under execution on the basis of ProQR’s business plan and budget.
(e) Use of critical estimates and judgements
In preparing these consolidated financial statements, management has made judgements, estimates and assumptions that affect the application of accounting policies and the reported amounts of assets, liabilities, income and expenses. Actual results may differ from these estimates.
Estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period or in the period of the revision and future periods if the revision affects both current and future periods.
Information about assumptions and estimation uncertainties that may have a significant risk of resulting in a material adjustment is included below:
F-8
(i) Revenue recognition for the Eli Lilly research and collaboration agreement
a. Identification of the performance obligation
Note 17 to the financial statements included elsewhere in this Annual Report describes the Company’s original research and collaboration agreement with Eli Lilly and Company, and the amended and restated research and collaboration agreement (collectively, the “Collaboration agreement”). Under the Collaboration agreement, ProQR provides Eli Lilly with a license (with a right to sub-license) to exploit compounds resulting from the collaboration. A significant amount of judgement is required to determine whether the license is distinct from the other promises in the contract. The license was concluded not to be distinct from the other promises in the contract based on the following considerations:
| ● | the license has no stand-alone value to Eli Lilly without the Company being involved in the research and development collaboration, and; |
| ● | there are significant interdependencies between the license and the research and development services to be provided by the Company. |
b. Determining the timing of satisfaction of performance obligations
Under the Collaboration agreement, the Company recognizes revenue over time, using an input method that estimates the satisfaction of the performance obligation as the percentage of labor hours incurred compared to the total estimated labor hours required to complete the promised services. As our estimate of the total labor hours required is dependent on the evolution of the research and development activities, it may be subject to change. If the progression and/or outcome of certain research and development activities would be different from the assumptions that were made during the preparation of these financial statements, this could lead to material adjustments to the total estimated labor hours, which might result in a reallocation of revenue between current and future periods. Our total deferred revenue balance related to this Eli Lilly performance obligation amounts to €
c. Determining the transaction price
The Company applied judgement to determine whether the equity investments made by Eli Lilly in ProQR are part of the transaction price for the Collaboration agreement. The Company concluded that the differences between the prices that Eli Lilly paid for the shares and the ProQR stock closing prices on the days of entering into the equity investment agreements arose because of the Company’s existing obligations to deliver research and development services to Eli Lilly under the terms of the Collaboration agreement. Therefore, the above differences between the closing share prices on the agreement effective dates and the equity investment prices paid by Lilly are considered to be part of the transaction price of the contract and are initially allocated to deferred revenue.
The contract also includes variable consideration, but
(ii) Research and development expenditures
Research expenditures are reflected in the income statement. Development expenses are currently also reflected in the income statement because the criteria for capitalization are not met. At each balance sheet date, the Company estimates the level of service performed by the vendors and the associated costs incurred for the services performed.
Although we do not expect the estimates to be materially different from amounts actually incurred, the understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and could result in reporting amounts that are too high or too low in any particular period.
F-9
(f) Changes in accounting policies
The following standards, amendments to standards and interpretations became effective for annual reporting periods beginning on or after January 1, 2023:
| ● | IFRS 17 Insurance contracts: This standard is aimed at reporting entities who are insurers. |
| ● | IAS 12 Income taxes: Amendments clarifying how companies account for deferred tax on transactions such as leases and decommissioning obligations. |
| ● | IAS 8: Accounting policies, changes in accounting estimates and errors: Amendments enabling entities to distinguish between accounting policies and accounting estimates. |
| ● | IAS 1 Presentation of financial statements: Amendments clarifying which accounting policies and accounting estimates preparers need to disclose. |
| ● | IFRS 16 Leases: Amendments relating to sale and leaseback transactions. |
In Note 23. Income taxes, we disclosed the impact of the amendments to IAS 12. None of the other new standards, amendments to standards and interpretations had a material impact on our financial statements. No changes in accounting policies occurred in 2023.
3. Significant Accounting Policies
The Company has consistently applied the following accounting policies to all periods presented in these consolidated financial statements.
(a) Basis of consolidation
(i) Subsidiaries
Subsidiaries are entities controlled by the Company. The Company controls an entity when it has power over the entity, is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power over the entity. The Company reassesses whether or not it controls an entity if facts and circumstances indicate that there are changes to one or more of these elements. The financial statements of subsidiaries are included in the consolidated financial statements from the date on which control commences until the date on which control ceases.
(ii) Non-controlling interests (“NCI”)
NCI are measured at their proportionate share of the acquiree's identifiable net assets at the acquisition date. Changes in the Company's interest in a subsidiary that do not result in a loss of control are accounted for as equity transactions.
(iii) Loss of control
When the Company loses control over a subsidiary, it derecognizes the assets and liabilities of the subsidiary, and any non-controlling interests and other components of equity. Any resulting gain or loss is recognized in profit or loss. Any interest retained in the former subsidiary is measured at fair value when control is lost.
(iv) Transactions eliminated on consolidation
Intra-group balances and transactions, and any unrealized income and expenses arising from intra-group transactions, are eliminated. Unrealized gains arising from transactions with equity-accounted investees are eliminated against the investment to the extent of the Company’s interest in the investee. Unrealized losses are eliminated in the same way as unrealized gains, but only to the extent that there is no evidence of impairment.
F-10
(v) Associates
Associates are entities over which the Company has significant influence. Significant influence is the power to participate in the financial and operating policy decisions of the investee but is not control or joint control over those policies.
Investments in associates are accounted for in the consolidated financial statements using the equity method of accounting. Equity accounting involves recording the investment in associates initially at cost, and recognizing the Company’s share of the post-acquisition results of associates in the consolidated income statement and the Company’s share of post-acquisition other comprehensive income in consolidated other comprehensive income. The cumulative post-acquisition movements are adjusted against the carrying amount of the investments in associates in the consolidated statement of financial position.
When the Company’s share of losses in an associate equals or exceeds its interest in the associate, the Company does not recognize further losses unless it has incurred or guaranteed obligations in respect of the associate.
(b) Classes of financial instruments
Financial instruments are both primary financial instruments, such as receivables and payables, and financial derivatives. For the Company’s primary financial instruments, reference is made to the treatment per the corresponding balance sheet item.
Financial derivatives are valued at fair value. Upon first recognition, financial derivatives are recognized at fair value and then revalued as at balance sheet date. Changes in the fair value of derivatives are generally recognized in profit or loss. If the Company is involved with hybrid contracts, the Company applies the following with regard to the embedded derivatives in the hybrid contract. Embedded derivatives are separated from the host contract and accounted for separately if the host contract is not a financial asset and the following criteria are met:
| ● | the economic characteristics and risk of the embedded derivative are not closely related to the economic characteristics and risks of the host contract; |
| ● | a separate instrument with the same terms as the embedded derivative would meet the definition of a derivate; and |
| ● | the hybrid contract is not measured at fair value with changes in fair value recognized in profit or loss. |
If an embedded derivative is separated from the hybrid contract, the host contract is accounted for in accordance with the determined policies for such a contract. The embedded derivative is accounted for in accordance with the Company’s principles for the applicable derivatives.
(c) Foreign currencies
(i) Foreign currency transactions
Foreign currency transactions are translated into the functional currency using the exchange rates prevailing at the dates of the transactions.
F-11
Monetary assets and liabilities denominated in foreign currencies are translated into the functional currency at the exchange rate at the reporting date. Non-monetary assets and liabilities denominated in foreign currencies that are measured at fair value are translated into the functional currency at the exchange rate when the fair value was determined. Foreign currency differences are generally recognized in profit or loss. Non-monetary items that are measured based on historical cost in a foreign currency are translated at the exchange rate prevailing at the date of the transaction.
(ii) Foreign operations
The assets and liabilities of foreign operations are translated into euro at exchange rates at the reporting date. The income and expenses of foreign operations are translated into euros at the exchange rates at the dates of the transactions. Foreign currency differences are recognized in OCI and accumulated in the translation reserve, except to the extent that the translation difference is allocated to NCI.
(d) Revenue
Revenues to date have consisted principally of non-refundable upfront fees and research and development service fees in connection with collaboration and license agreements. The Company recognizes revenue when its customers obtain control of promised goods or services, in an amount that reflects the consideration that the Company expects to receive in exchange for those goods and services. Revenue is recognized for agreements that are in scope of IFRS 15 Revenue from contracts with customers, based on the following five steps:
| (i) | Identify the contract |
The Company entered into collaboration and license agreements in which the Company licenses its intellectual property and/or provides research and development services. These arrangements include upfront payments, milestone payments based on clinical and regulatory criteria, research and development service fees and future sales-based milestones and sales-based royalties. In some cases, concurrently with the collaboration and license agreements, the Company enters into share purchase agreements with the customer. If this is the case, the Company analyzes whether the criteria to combine contracts, as set out by IFRS 15, are met.
(ii) Identify performance obligations
Contracts with customers can have one or more distinct performance obligations under IFRS 15. Identifying the performance obligations is based on an assessment of whether the promises in an agreement are capable of being distinct and are distinct from the other promises to transfer goods and/or services in the context of the contract. The Company assessed that there is one performance obligation in our material ongoing collaboration and license agreements, for the transfer of a license combined with performance of research and development services.
This is because the Company considers the two obligations cannot be distinct in the context of the contract as the licenses have no stand-alone value without the Company being involved in the research and development collaboration and that there is interdependence between the license and the research and development services to be provided.
(iii) Determine the transaction price
Our research and collaboration agreements include non-refundable upfront payments; equity components; milestone payments, the receipt of which is dependent upon the achievement of certain clinical, regulatory or commercial milestones; royalties on sales and research and development service fees.
| a. | Non-refundable upfront payments or license fees |
If the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Company recognizes revenue from non-refundable upfront fees allocated to this license at the point in time the license is transferred to the customer and the customer has the right to use the license.
F-12
For all our material ongoing research and collaboration agreements, the Company considers the performance obligations related to the transfer of the license as not distinct from the other promises to transfer goods and/or services; the Company uses judgement to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time. If over time, revenue is then recognized based on a pattern that best reflects the transfer of control of the service to the customer.
| b. | Milestone payments other than sales-based milestones |
A milestone payment, being a variable consideration, is only included in the transaction price to the extent it is highly probable that a significant reversal in the amount of cumulative revenue recognition will not occur when the uncertainty associated with the variable consideration is subsequently resolved. The Company estimates the amount to be included in the transaction price upon achievement of the milestone event. The transaction price is then allocated to each performance obligation on a stand-alone selling price basis, for which the Company recognizes revenue as or when the performance obligations under the contract are satisfied. At the end of each reporting period, the Company re-evaluates the probability of achievement of such milestones and any related constraint, and, if necessary, adjusts the estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect revenue and earnings in the period of adjustment.
| c. | Research and development service fees |
Our collaboration and license agreements may include reimbursement for research and development services. R&D services are performed and satisfied over time because the customer simultaneously receives and consumes the benefits provided by us. Revenue associated with such R&D service fees is then recognized based on a pattern that best reflects the transfer of control of the service to the customer.
| d. | Sales based milestone payments and royalties |
Our material collaboration and license agreements include sales-based royalties, including commercial milestone payments based on the level of sales. The Company concluded that the licenses are not the predominant items to which the royalties and commercial milestone payments relate. Related revenue will be recognized as the subsequent underlying sales occur.
(iv) Allocate the transaction price
An entity shall allocate the transaction price to each performance obligation identified in a contract on a relative stand-alone selling price basis. As our collaboration and license agreements only contain
(v) Recognize revenue
Revenue is recognized when the customer obtains control of the goods and/or services as provided in the research and collaboration agreements. Control can be transferred over time or at a point in time, which results in the recognition of revenue either over time or at a point in time.
Our research and collaboration agreements only contain
The recognition of revenue over time is based on a pattern that best reflects the satisfaction of the related performance obligation, applying the input method. The input method estimates the satisfaction of the performance obligation as the percentage of labor hours incurred compared to the total estimated labor hours required to complete the promised services.
F-13
(e) Other income
Other income includes amounts earned from third parties and are recognized when earned in accordance with the substance and under the terms of the related agreements and when it is probable that the economic benefits associated with the transaction will flow to the Company and the amount of the income can be measured reliably. The grants are recognized in other income on a systematic basis over the period the Company recognizes as expenses the related costs for which the grants are expected to compensate.
(f) Government grants — WBSO
The WBSO (“afdrachtvermindering speur- en ontwikkelingswerk”) is a Dutch fiscal facility that provides subsidies to companies, knowledge centers and self-employed people who perform research and development activities (as defined in “the WBSO Act”). Under this Act, a contribution is paid towards the labor costs of employees directly involved in research and development. The contribution is in the form of a reduction of payroll taxes and social security contributions recognized on a net basis within the labor costs. This reduction of payroll taxes and social security contributions is classified under research and developments costs.
(Government) Grant income is not recognized until there is reasonable assurance that the Company will comply with the conditions attached to them. (Government) Grants are recognized in profit or loss on a systematic basis over the period the Company recognizes as expenses the related costs for which the grants are intended to compensate.
(g) Employee benefits
(i) Short-term employee benefits
Short-term employee benefits are expensed as the related service is provided. A liability is recognized for the amount expected to be paid if the Company has a present legal or constructive obligation to pay this amount as a result of past service provided by the employee and the obligation can be estimated reliably.
(ii) Share-based payment transactions
The grant-date fair value of equity-settled share-based payment awards granted to employees is generally recognized as an expense, with a corresponding increase in equity, over the vesting period of the awards. The amount recognized as an expense is adjusted to reflect the number of awards for which the related service and non-market performance conditions are expected to be met, such that the amount ultimately recognized is based on the number of awards that meet the related service conditions at the vesting date. For share-based payment awards with non-vesting conditions, the grant-date fair value of the share-based payment is measured to reflect such conditions and there is no true-up for differences between expected and actual outcomes.
(iii) Pension obligations
The Company operates defined contribution pension plans for all employees funded through payments to insurance companies. The Company has no legal or constructive obligation to pay further contributions once the contributions have been paid. The contributions are recognized as employee benefit expense when employees have rendered the service entitling them to the contributions. Prepaid contributions are recognized as an asset to the extent that a cash refund or a reduction in the future payments is available.
(h) Taxation
Income tax expense represents the sum of the tax currently payable and deferred tax. It is recognized in profit or loss except to the extent that it relates to a business combination, or items recognized directly in equity or in OCI.
F-14
(i) Current tax
The tax currently payable is based on taxable profit for the year. Taxable profit differs from profit as reported in the income statement because of items of income or expense that are taxable or deductible in other years and items that are never taxable or deductible. The Company’s liability for current tax is calculated using tax rates that have been enacted or substantively enacted by the end of the reporting period.
(ii) Deferred tax
Deferred tax is recognized on differences between the carrying amounts of assets and liabilities in the financial statements and the corresponding tax bases used in the computation of taxable profit.
The carrying amount of deferred tax assets is reviewed at the end of each reporting period and reduced to the extent that it is no longer probable that sufficient taxable profits will be available to allow all or part of the asset to be recovered. Since the Company does not expect to be profitable in the foreseeable future, its deferred tax assets are valued at
Deferred tax assets and liabilities are measured at the tax rates that are expected to apply in the period in which the liability is settled or the asset realized, based on tax rates (and tax laws) that have been enacted or substantively enacted by the end of the reporting period. The measurement of deferred tax liabilities and assets reflects the tax consequences that would follow from the manner in which the Company expects, at the end of the reporting period, to recover or settle the carrying amount of its assets and liabilities.
Deferred tax assets and liabilities are offset when there is a legally enforceable right to offset current tax assets against current tax liabilities and when they relate to income taxes levied by the same taxation authority and the Company intends to settle its current tax assets and liabilities on a net basis.
(i) Property, plant and equipment
(i) Recognition and measurement
Items of property, plant and equipment are measured at cost less accumulated depreciation and any accumulated impairment losses. If significant parts of an item of property, plant and equipment have different useful lives, then they are accounted for as separate items (major components) of property, plant and equipment. Any gain or loss on disposal of an item of property, plant and equipment is recognized in profit or loss.
(ii) Depreciation
Depreciation is calculated to write off the cost of items of property, plant and equipment less their estimated residual values using the straight-line method over their estimated useful lives and is recognized in profit or loss. Right-of-use assets are depreciated over the shorter of the lease term and their useful lives unless it is reasonably certain that the Company will obtain ownership by the end of the lease term.
The estimated useful lives of property, plant and equipment for current and comparative periods are as follows:
● buildings and leasehold improvements: |
| years | |
● laboratory equipment: |
| years | |
● other: |
| years |
Depreciation methods, useful lives and residual values are reviewed at each reporting date and adjusted if appropriate.
F-15
(j) Intangible assets
Expenditure on research activities is recognized as an expense in the period in which it is incurred. An internally-generated intangible asset arising from development (or from the development phase of an internal project) is recognized if, and only if, all of the following have been demonstrated:
| ● | the technical feasibility of completing the intangible asset so that it will be available for use or sale; |
| ● | the intention to complete the intangible asset and use or sell it; |
| ● | the ability to use or sell the intangible asset; |
| ● | how the intangible asset will generate probable future economic benefits; |
| ● | the availability of adequate technical, financial and other resources to complete the development and to use |
| ● | or sell the intangible asset; and |
| ● | the ability to measure reliably the expenditure attributable to the intangible asset during its development. |
The amount initially recognized for internally-generated intangible assets is the sum of the expenditure incurred from the date when the intangible asset first meets the recognition criteria listed above. Where no internally generated intangible asset can be recognized, development expenditures are recognized in the consolidated statements of profit and loss and other comprehensive income in the period in which they are incurred.
Due to uncertainties inherent to the development and registration with the relevant healthcare authorities of its products, the Company estimates that the conditions for capitalization are not met until the regulatory procedures required by such healthcare authorities have been finalized. The Company currently does not own products that have been approved by the relevant healthcare authorities and this has resulted in all development costs being recognized as an expense in the period in which they are incurred.
(k) Impairment of assets
At the end of each reporting period, the Company reviews the carrying amounts of its non-current assets, including right-of-use assets, to determine whether there is any indication that those assets have suffered an impairment loss. If any such indication exists, the recoverable amount of the asset is estimated in order to determine the extent of the impairment loss (if any). Where it is not possible to estimate the recoverable amount of an individual asset, the Company estimates the recoverable amount of the cash-generating unit to which the asset belongs. Where a reasonable and consistent basis of allocation can be identified, corporate assets are also allocated to individual cash-generating units, or otherwise they are allocated to the smallest group of cash-generating units for which a reasonable and consistent allocation basis can be identified.
The recoverable amount is the higher of fair value less costs of disposal and value in use. In assessing value in use, the estimated future cash flows are discounted to their present value using a pre-tax discount rate that reflects current market assessments of the time value of money and the risks specific to the asset for which the estimates of future cash flows have not been adjusted.
If the recoverable amount of an asset (or cash-generating unit) is estimated to be less than its carrying amount, the carrying amount of the asset (or cash-generating unit) is reduced to its recoverable amount. An impairment loss is recognized immediately in profit or loss.
Where an impairment loss subsequently reverses, the carrying amount of the asset (or cash-generating unit) is increased to the revised estimate of its recoverable amount, but so that the increased carrying amount does not exceed the carrying amount that would have been determined had no impairment loss been recognized for the asset (or cash-generating unit) in prior years. A reversal of an impairment loss is recognized immediately in profit or loss, unless the relevant asset is carried at a revalued amount, in which case the reversal of the impairment loss is treated as a revaluation increase.
F-16
(l) Financial assets
All financial assets are recognized and derecognized on the trade date where the purchase or sale of a financial asset is under a contract whose terms require delivery of the financial asset within the timeframe established by the market concerned, and are initially measured at fair value and subsequently measured at amortized cost or fair value on the basis of the entity’s business model for managing the financial assets and the contractual cash flow characteristics of the financial assets.
Specifically:
| ● | debt instruments that are held within a business model whose objective is to collect the contractual cash flows, and that have contractual cash flows that are solely payments of principal and interest on the principal amount outstanding, are measured subsequently at amortized cost, and |
| ● | all other debt investments and equity investments are measured subsequently at fair value through profit or loss (“FVTPL”). |
The Company applies the IFRS 9 simplified approach to measuring expected credit losses which uses a lifetime expected loss allowance for all trade receivables. To measure the expected credit losses, trade receivables have been grouped based on shared credit risk characteristics and the days past due. Trade receivables are written off when there is no reasonable expectation of recovery. Indicators that there is no reasonable expectation of recovery include, amongst others, the failure of a debtor to engage in a repayment plan with the group, and a failure to make contractual payments for a period of greater than 120 days past due. Impairment losses on trade receivables and contract assets are presented as net impairment losses within operating profit. Subsequent recoveries of amounts previously written off are credited against the same line item.
The Company derecognizes a financial asset when the contractual rights to the cash flows from the asset expire, or the Company transfers the right to receive the contractual cash flows on the financial asset in a transaction in which substantially all the risks and rewards of ownership of the financial asset are transferred.
(m) Cash and cash equivalents
Cash and cash equivalents include cash on hand and all highly liquid investments with original maturities of three months or less that are readily convertible to a known amount of cash and bear an insignificant risk of change in value.
(n) Financial liabilities and equity instruments
Debt and equity instruments are classified as either financial liabilities or as equity in accordance with the substance of the contractual arrangement.
Equity instruments
An equity instrument is any contract that evidences a residual interest in the assets of an entity after deducting all of its liabilities. Equity instruments issued by the Company are recognized at the proceeds received, net of direct issue costs.
Compound financial instruments
Compound financial instruments issued by the Company comprise convertible notes denominated in euro that can be converted to share capital at the option of the holder, when the number of shares to be issued is fixed and does not vary with changes in fair value.
F-17
The component parts of convertible loan notes issued by the Group are classified separately as financial liabilities and equity in accordance with the substance of the contractual arrangements and the definitions of a financial liability and an equity instrument. A conversion option that will be settled by the exchange of a fixed amount of cash or another financial asset for a fixed number of the Company’s own equity instruments is an equity instrument. At the date of issue, the fair value of the liability component is estimated using the prevailing market interest rate for a similar non-convertible instrument. This amount is recorded as a liability on an amortized cost basis using the effective interest method until extinguished upon conversion or at the instrument’s maturity date.
The conversion option classified as equity is determined by deducting the amount of the liability component from the fair value of the compound instrument as a whole. This is recognized and included in equity, net of income tax effects, and is not subsequently remeasured. In addition, the conversion option classified as equity will remain in equity until the conversion option is exercised, in which case, the balance recognized in equity will be transferred to share premium. Where the conversion option remains unexercised at the maturity date of the convertible loan note, the balance recognized in equity will be transferred to accumulated losses. No gain or loss is recognized in profit or loss upon conversion or expiration of the conversion option.
Transaction costs that relate to the issue of the convertible loan notes are allocated to the liability and equity components in proportion to the allocation of the gross proceeds. Transaction costs relating to the equity component are recognized directly in equity. Transaction costs relating to the liability component are included in the carrying amount of the liability component and are amortized over the lives of the convertible loan notes using the effective interest method.
Interest related to the financial liability is recognized in profit or loss.
Financial liabilities at fair value through profit or loss
Financial liabilities held for trading are classified as at fair value through profit or loss. A financial liability is classified as held for trading if it is a derivative (except for a derivative that is a financial guarantee contract or a designated and effective hedging instrument).
Financial liabilities at FVTPL are measured at fair value, with any gains or losses arising on changes in fair value recognized in profit or loss. The net gain or loss recognized is included in the ‘results related to financial liabilities measured at fair value through profit or loss’ line item in profit or loss.
Fair value is determined in the manner described in Note 5.
Other financial liabilities
Other financial liabilities, including borrowings, are initially measured at fair value, net of transaction costs incurred, and are subsequently measured at amortized cost using the effective interest method, with interest expense recognized on an effective yield basis.
The effective interest method is a method of calculating the amortized cost of a financial liability and of allocating interest expense over the relevant period. The effective interest rate is the rate that exactly discounts estimated future cash payments through the expected life of the financial liability, or, where appropriate, a shorter period.
Borrowings and other financial liabilities are classified as ‘non-current liabilities,’ other than liabilities with maturities up to one year, which are classified as “current liabilities”.
The Company derecognizes financial liabilities when the liability is discharged, cancelled or expired. For all financial liabilities, the fair value approximates its carrying amount.
F-18
Offsetting
Financial assets and financial liabilities are offset and the net amount presented in the statement of financial position when, and only when, the Company currently has a legally enforceable right to set off the amounts and it intends either to settle them on a net basis or to realize the asset and settle the liability simultaneously.
(o) Leases
The Company assesses whether a contract is or contains a lease when it obtains the right to control the use of an identified asset for a period of time, in exchange for consideration. The Company recognizes a right-of-use asset and a corresponding lease liability with respect to all lease arrangements in which it is the lessee, except for short-term leases (defined as leases with a lease term of 12 months or less) and leases of low value assets (such as tablets and personal computers, small items of office furniture and telephones). For these leases, the Company recognizes the lease payments in operating costs on a straight-line basis over the term of the lease unless another systematic basis is more representative of the time pattern in which economic benefits from the leased assets are consumed.
The lease liability is initially measured at the present value of the lease payments that are not paid at the commencement date, discounted by using the interest rate implicit in the lease. When the interest rate implicit in the lease cannot be readily determined, the Company uses its incremental borrowing rate.
Lease payments included in the measurement of the lease liability comprise:
| ● | Fixed lease payments (including in-substance fixed payments), less any lease incentives receivable; |
| ● | Variable lease payments that depend on an index or rate, initially measured using the index or rate at the commencement date; |
| ● | The amount expected to be payable by the Company under residual value guarantees; |
| ● | The exercise price of purchase options, if the Company is reasonably certain to exercise the options; and |
| ● | Payments of penalties for terminating the lease, if the lease term reflects the exercise of an option to terminate the lease. |
The lease liability is presented as a separate line in the consolidated statement of financial position. In the cash flow statement, repayments of the principal portion of the lease liability are included in financing activities. Payments relating to the interest component of the lease liability are included in operating activities. Short-term lease payments and payments for leases of low-value assets are included in operating activities.
The lease liability is subsequently measured by increasing the carrying amount to reflect interest on the lease liability (using the effective interest method) and by reducing the carrying amount to reflect the lease payments made.
The Company remeasures the lease liability (and makes a corresponding adjustment to the related right-of-use asset) whenever:
| ● | The lease term has changed or there is a significant event or change in circumstances resulting in a change in the assessment of exercise of a purchase option, in which case the lease liability is remeasured by discounting the revised lease payments using a revised discount rate |
F-19
| ● | The lease payments change due to changes in an index or rate or a change in expected payment under a guaranteed residual value, in which cases the lease liability is remeasured by discounting the revised lease payments using an unchanged discount rate (unless the lease payments change is due to a change in a floating interest rate, in which case a revised discount rate is used) |
| ● | A lease contract is modified and the lease modification is not accounted for as a separate lease, in which case the lease liability is remeasured based on the lease term of the modified lease by discounting the revised lease payments using a revised discount rate at the effective date of the modification |
The right-of-use asset comprises the initial measurement of the corresponding lease liability, lease payments made at or before the commencement day, less any lease incentives received and any initial direct costs. It is subsequently measured at cost less accumulated depreciation and impairment losses.
Whenever the Company incurs an obligation for costs to dismantle and remove a leased asset, restore the site on which it is located or restore the underlying asset to the condition required by the terms and conditions of the lease, a provision is recognized and measured under IAS 37. To the extent that the costs relate to a right-of-use asset, the costs are included in the related right-of-use asset, unless those costs are incurred to produce inventories.
Right-of-use assets are depreciated over the shorter period of lease term and useful life of the underlying asset. If a lease transfers ownership of the underlying asset or the cost of the right-of-use asset reflects that the Company expects to exercise a purchase option, the related right-of-use asset is depreciated over the useful life of the underlying asset. The depreciation starts at the commencement date of the lease.
The right-of-use asset is presented under Property, Plant and Equipment in the consolidated statement of financial position, in the category Buildings and leasehold improvements.
As a practical expedient, IFRS 16 permits a lessee not to separate non-lease components, and instead account for any lease and associated non-lease components as a single arrangement. The Company has used this practical expedient.
(p) Non-current assets held for sale
Non-current assets (and disposal groups) classified as held for sale are measured at the lower of carrying amount and fair value less costs to sell.
Non-current assets and disposal groups are classified as held for sale if their carrying amount will be recovered through a sale transaction rather than through continuing use. This condition is regarded as met only when the sale is highly probable and the asset (or disposal group) is available for immediate sale in its present condition. Management must be committed to the sale which should be expected to qualify for recognition as a completed sale within one year from the date of classification.
When the Company is committed to a sale plan involving loss of control of a subsidiary, all of the assets and liabilities of that subsidiary are classified as held for sale when the criteria described above are met, regardless of whether the Company will retain a non-controlling interest in its former subsidiary after the sale. When the Company is committed to a sale plan involving disposal of an investment in an associate or, a portion of an investment in an associate, the investment, or the portion of the investment in the associate, that will be disposed of is classified as held for sale when the criteria described above are met. The Company then ceases to apply the equity method in relation to the portion that is classified as held for sale. Any retained portion of an investment in an associate that has not been classified as held for sale continues to be accounted for using the equity method.
F-20
4. New standards and interpretations not yet adopted
A number of new standards, amendments to standards and interpretations are effective for annual periods beginning after January 1, 2024 and have not been applied in preparing these consolidated financial statements. There are no standards that are not yet effective and that would be expected to have a material impact on the Company in the current or future reporting periods and on foreseeable future transactions. The Company does not plan to adopt these standards early.
5. Financial Risk Management
5.1 Financial risk factors
The Company’s activities expose it to a variety of financial risks: market risk (including currency risk, interest rate risk and price risk), credit risk and liquidity risk. The Company’s overall financial risk management seeks to minimize potential adverse effects resulting from unpredictability of financial markets on the Company’s financial performance.
Financial risk management is carried out by the finance department. The finance department identifies and evaluates financial risks and proposes mitigating actions if deemed appropriate.
(a) Market risk
Market risk is the risk that changes in market prices – such as foreign exchange rates, interest rates and equity prices – will affect the Company’s income or the value of its holdings of financial instruments. The objective of market risk management is to manage and control market risk exposures within acceptable parameters, while optimizing the return.
Foreign exchange risk
Foreign exchange risk arises from future commercial transactions and recognized assets and liabilities in foreign currencies, primarily with respect to the U.S. dollar. The Company has an exposure associated with the time delay between entering into a contract, budget or forecast and the realization thereof. The Company operates a foreign exchange policy to manage the foreign exchange risk against the functional currency based on the Company’s cash balances and the projected future spend per major currency.
At year-end, a substantial amount of our cash balances are denominated in U.S. Dollars. This amount reflects our current expectation of future expenditure in U.S. dollars.
At December 31, 2023 our net position of financial instruments denominated in U.S. dollars was a net liability of €
A reasonably possible weakening of the U.S. dollar by
Price risk
The market prices for the production of preclinical materials and services as well as external contracted research may vary over time. Currently, the commercial prices of any of the Company’s future product candidates is uncertain. When product candidates approach the regulatory approval date or potential regulatory approval date, the uncertainty of potential sales prices decreases. The Company is not exposed to commodity price risk.
F-21
Furthermore, the Company does not hold investments designated for sale and is therefore not exposed to equity securities price risk.
Cash flow and fair value interest rate risk
The Company’s interest rate risk arises from current accounts and deposits and the sensitivity analysis below has been determined based on the exposure to interest rates on these short-term maturity primary financial instruments.
A 10 percent increase or decrease on actual interest rate is used when reporting interest rate risk internally to key management personnel and represents management’s assessment of the reasonably possible change in interest rates.
As of December 31, 2023, if interest rates had been
The Company’s exposure to interest rate risks on loans and leases is limited due to the use of fixed interest rates. The Company has a loan with a fixed interest rate, totaling €
(b) Credit risk
Credit risk represents the risk of financial loss caused by default of the counterparty. The Company has no large receivables balances with external parties outside of cash and cash equivalents. Our cash management policy is focused on preserving capital, providing liquidity for operations and optimizing yield while accepting limited risk (Short-term credit ratings must be rated A-1/P-1/F1 at a minimum by at least one of the Nationally Recognized Statistical Rating Organizations (“NRSROs”) specifically Moody’s, Standard & Poor’s or Fitch. Long-term credit rating must be rated A2 or A at a minimum by at least one NRSRO). As of December 31, 2023, the Company is in compliance with its cash management policy.
At December 31, 2023 and December 31, 2022, all of our cash and cash equivalents were held at
There are
(c) Liquidity risk
Liquidity risk represents the risk that an entity will encounter difficulty in meeting obligations associated with its financial liabilities. Prudent liquidity risk management implies ensuring sufficient availability of cash resources for funding of operations and planning to raise cash if and when needed, either through issue of shares or through credit facilities. Management monitors rolling forecasts of the Company’s liquidity reserve on the basis of expected cash flow.
F-22
The table below analyzes ProQR’s undiscounted liabilities into relevant maturity groupings based on the remaining period at year-end until the contractual maturity date:
|
| Less than |
| Between 1 |
| Between 2 |
| |
1 year | and 2 years | and 5 years | Over 5 years | |||||
(€ in thousands) | ||||||||
At December 31, 2023 |
|
|
|
|
|
|
|
|
Borrowings |
| — | | — | — | |||
Lease liabilities | | | | | ||||
Trade payables and other payables |
| | — | — |
| — | ||
Total |
| |
| |
| |
| |
At December 31, 2022 |
|
|
|
|
|
|
|
|
Borrowings |
| | | | — | |||
Lease liabilities | | | | | ||||
Trade payables and other payables |
| | — | — |
| — | ||
Total |
| |
| |
| |
| |
Our future capital requirements and the period for which our existing resources will support our operations may vary significantly from what we expect. Our monthly spending levels will vary based on new and ongoing development and corporate activities. Because the length of time and activities associated with successful development of our product candidates is highly uncertain, we are unable to estimate the actual funds we will require for development of our product candidates.
5.2 Capital risk management
The Company’s objectives when managing capital are to safeguard the Company’s ability to continue as a going concern in order to provide returns for shareholders, benefits for other stakeholders and to maintain an optimal capital structure to reduce the cost of capital.
In order to maintain or adjust the capital structure, the Company may adjust the amount of dividends paid to shareholders (although at this time the Company does not have retained earnings and is therefore currently unable to pay dividends), return capital to shareholders, issue new shares or sell assets to reduce debt.
The total amount of equity as recorded on the balance sheet is managed as capital by the Company.
5.3 Fair value measurement
For financial instruments that are measured on the balance sheet at fair value, IFRS 13 requires disclosure of fair value measurements by level of the following fair value measurement hierarchy:
| ● | quoted prices (unadjusted) in active markets for identical assets or liabilities (level 1); |
| ● | inputs other than quoted prices included within level 1 that are observable for the asset or liability, either directly (that is, as prices) or indirectly (that is, derived from prices) (level 2); and |
| ● | inputs for the asset or liability that are not based on observable market data (that is, unobservable inputs) (level 3). |
F-23
Fair value of financial assets and liabilities that are measured at fair value on a recurring basis
Some of the Company’s financial assets and liabilities are measured at fair value at the end of each reporting period. The following table gives information about how the fair values of these financial assets and liabilities are determined (in particular, the valuation technique and inputs used).
Relationship and | ||||||
sensitivity of | ||||||
|
| Valuation technique |
| Significant unobservable |
| Significant unobservable |
Financial liabilities | and key inputs | inputs | inputs to fair value | |||
|
|
|
|
| ||
Investment in Phoenicis Therapeutics, Inc | Market comparison technique: The valuation model is based on market multiples derived from quoted prices of companies comparable to the investee, adjusted for the effect of the non-marketability of the equity securities, and the result of the investee. The estimate is adjusted for the net debt of the investee. | Adjusted market-multiple | The estimated fair value would increase (decrease) if the adjusted market-multiple were higher (lower). | |||
Investment in Yarrow Biotechnology, Inc. | Market comparison technique: The valuation model is based on market multiples derived from quoted prices of companies comparable to the investee, adjusted for the effect of the non-marketability of the equity securities, and the result of the investee. The estimate is adjusted for the net debt of the investee. | Adjusted market-multiple | The estimated fair value would increase (decrease) if the adjusted market-multiple were higher (lower). | |||
Warrants and conversion options |
| Black-Scholes model. The following variables were taken into consideration: current underlying price of the Company's shares, options strike price, expected life, historical volatility of ProQR share returns over a period equal to the expected life, risk-free rate: based on the US Treasury yield curve rates per the valuation date (interpolated) for the expected life. | Not applicable | Not applicable |
The investments in Phoenicis Therapeutics, Inc and Yarrow Biotechnology, Inc are measured using valuation methods based on so-called Level 3 inputs. Level 3 inputs are unobservable inputs. Changing one or more of the unobservable inputs to reflect reasonably possible alternative assumptions would not significantly change the fair value determined for Phoenicis Therapeutics, Inc and Yarrow Biotechnology, Inc.
Warrants are measured using valuation methods based on so-called Level 2 inputs. Level 2 inputs are inputs other than quoted prices that are observable for the liability, either directly or indirectly.
The carrying amount of all financial assets and financial liabilities is a reasonable approximation of the fair value and therefore information about the fair values of each class has not been disclosed.
Share options and restricted stock units (“RSUs”) granted to employees and consultants are measured at the fair value of the equity instruments granted. The fair value of options is determined through the use of an option-pricing model considering, among others, the following variables:
| ● | the exercise price of the option; |
| ● | the expected life of the option; |
| ● | the current value of the underlying shares; |
F-24
| ● | the expected volatility of the share price; |
| ● | the dividends expected on the shares; and |
| ● | the risk-free interest rate for the life of the option. |
6. Segment Information
The Company operates in
Revenues are generated from external customers whose main registered offices are all geographically located in the United States. Substantially all non-current assets of the Company are located in the Netherlands. The amounts provided to the management board with respect to total assets and liabilities are measured in a manner consistent with that of the financial statements.
F-25
7. Property, Plant and Equipment
|
| Buildings and |
|
|
| |||
leasehold | Laboratory | |||||||
improvements | equipment | Other | Total | |||||
(€ in thousands) | (€ in thousands) | (€ in thousands) | (€ in thousands) | |||||
Balance at January 1, 2022 |
|
|
|
|
|
|
|
|
Cost |
| |
| |
| |
| |
Accumulated depreciation |
| ( |
| ( |
| ( |
| ( |
Carrying amount |
| |
| |
| |
| |
Additions |
| | | |
| | ||
Depreciation |
| ( | ( | ( |
| ( | ||
Effect of lease modification (Note 25) | | — | — | | ||||
Transfers | ( | | ( | — | ||||
Disposals |
| — | ( | — |
| ( | ||
Movement for the period |
| ( |
| |
| ( |
| ( |
Balance at December 31, 2022 |
|
|
|
|
|
|
|
|
Cost |
| |
| |
| |
| |
Accumulated depreciation |
| ( |
| ( |
| ( |
| ( |
Carrying amount |
| |
| |
| |
| |
Balance at January 1, 2023 | ||||||||
Cost |
| |
| |
| |
| |
Accumulated depreciation |
| ( |
| ( |
| ( |
| ( |
Carrying amount |
| |
| |
| |
| |
Additions |
| | | |
| | ||
Depreciation |
| ( | ( | ( |
| ( | ||
Effect of lease modification (Note 25) | | — | — | | ||||
Transfer | | ( | | — | ||||
Disposals - cost | — | ( | — | ( | ||||
Accumulated depreciation on disposals |
| — | | — |
| | ||
Movement for the period |
| ( |
| |
| |
| |
Balance at December 31, 2023 |
|
|
|
|
|
|
|
|
Cost |
| | | |
| | ||
Accumulated depreciation |
| ( | ( | ( |
| ( | ||
Carrying amount |
| |
| |
| |
| |
The depreciation charge for 2023 is included in research and development costs for an amount of €
Buildings and leasehold improvements include a right-of-use asset relating to the lease of our Leiden office and laboratory space, with a carrying amount of €
F-26
8. Investments in Associates
In May 2021, the Company obtained an
In October 2023, Gerard Platenburg, Chief Scientific Officer at ProQR, ended his term on Yarrow’s board of directors. From that moment onwards, ProQR no longer had significant influence over Yarrow. Yarrow was therefore derecognized as an associate and was accounted for as a financial asset, as disclosed in Note 9.
As the carrying amount of our investment in Yarrow was €
|
| Investment |
in associate | ||
(€ in thousands) | ||
Balance at January 1, 2022 | | |
Share of loss from continuing operations | ( | |
Balance at December 31, 2022 |
| — |
Derecognition of investment in associate (Yarrow Biotechnology, Inc.) | — | |
Balance at December 31, 2023 |
| — |
9. Investments in Financial Assets
Yarrow Biotechnology, Inc.
As disclosed in Note 8, Gerard Platenburg, Chief Scientific Officer at ProQR, ended his term on Yarrow’s board of directors in October 2023. From then on, ProQR no longer had significant influence over Yarrow. Yarrow was therefore derecognized as an associate and was accounted for as a financial asset and measured at fair value.
ProQR holds a
Phoenicis Therapeutics, Inc.
In May 2019, the Company acquired a non-controlling interest in Wings Therapeutics Inc. (“Wings”) as part of the
strategic spin out of its Dystrophic Epidermolysis Bullosa (“DEB”) activities. In January 2021, Wings merged into
Phoenicis Therapeutics Inc. (“Phoenicis”) by means of a non-cash transaction. Consequently, Wings ceased to exist, and the related investment was derecognized. In 2021, a gain on disposal of associate was recognized amounting to €
F-27
ProQR holds a
10. Other Taxes
|
| December 31, |
| December 31, |
2023 | 2022 | |||
| (€ in thousands) | |||
Value added tax |
| |
| |
| |
| | |
All receivables are considered short-term and due within one year.
11. Prepayments and Other Receivables
|
| December 31, |
| December 31, |
2023 | 2022 | |||
| (€ in thousands) | |||
Prepayments |
| |
| |
Eli Lilly up-front receivable | — | | ||
Other receivables |
| |
| |
| |
| | |
All receivables are considered short-term and due within one year. At December 31, 2023 and 2022, prepayments consisted principally of payments made by the Company for services not yet provided by vendors. At December 31, 2023 and 2022, other receivables consisted principally of deposits. Note 17 Revenue describes the transaction related to the Eli Lilly up-front receivable.
12. Cash and Cash Equivalents
|
| December 31, |
| December 31, |
2023 | 2022 | |||
| (€ in thousands) | |||
Cash at banks |
| |
| |
Deposits | | — | ||
| |
| | |
The cash at banks is at full disposal of the Company. Deposits are fixed for at most 3 month periods at a time.
F-28
13. Shareholders’ Equity
(a) Share capital
Number of shares 2023 | Number of shares 2022 | Number of shares 2021 | ||||
|
| Ordinary |
| Ordinary |
| Ordinary |
Balance at January 1 |
| |
| |
| |
Issued for cash |
| — |
| |
| |
Issued for services | — | — | | |||
Exercise of share options / vesting of RSUs |
| |
| |
| |
Treasury shares issued (transferred) |
| ( |
| ( |
| ( |
Balance at December 31 |
| |
| |
| |
The authorized share capital of the Company amounting to €
In April 2021, the Company consummated an underwritten public offering of
In September 2021, the Company issued
In November, 2021, the Company filed a shelf registration statement, which permitted: (a) the offering, issuance and sale by the Company of up to a maximum aggregate offering price of $
In December 2022, the Company issued
(b) Equity settled employee benefit reserve
The costs of share options and RSUs for employees, members of the Supervisory Board and members of the Management Board are recognized in the income statement, together with a corresponding increase in equity during the vesting period, taking into account (deferral of) corporate income taxes. The accumulated expense of share-based compensation recognized in the income statement is shown separately in the equity category ‘equity settled employee benefit reserve’ in the ‘statement of changes in equity’. On September 25, 2017, we established a Dutch foundation named Stichting Bewaarneming Aandelen ProQR for holding shares in trust for employees, members of the Management Board and members of the Supervisory Board of the Company and its group companies who from time to time could exercise options under the Company’s equity incentive plans.
(c) Translation reserve
The translation reserve comprises all foreign currency differences arising from the translation of the financial statements of foreign operations.
F-29
(d) Share options and restricted stock units
The Company operates an equity-settled share-based compensation plan which was introduced in 2013. Options and RSUs may be granted to employees, members of the Supervisory Board, members of the Management Board and consultants. The compensation expenses included in operating costs for this plan were €
Options granted under this stock option plan are exercisable once vested. Any vesting schedule may be attached to the granted options and RSUs. Typical vesting periods are:
| ● |
| ● |
| ● |
The options expire
The fair value of the options is estimated at the date of grant using the Black-Scholes option-pricing model, with on average the following assumptions:
| Options granted |
| Options granted |
| Options granted |
| |
in 2023 | in 2022 | in 2021 |
| ||||
Risk-free interest rate |
| | % | | % | | % |
Expected dividend yield |
| — | % | — | % | — | % |
Expected volatility |
| | % | | % | | % |
Expected life in years |
| | years | | years | | years |
The resulting weighted average grant date fair value of the options amounted to €
The fair value of RSUs is determined at the grant date by using the Company’s share price at the grant date. The resulting weighted average grant date fair value of the RSUs amounted to €
Movements in the number of options outstanding and their related weighted average exercise prices are as follows:
| 2023 | 2022 | 2021 | ||||||||||||
|
| Number of |
| Average |
| Number of |
| Average |
| Number of |
| Average | |||
options | exercise price | options | exercise price | options | exercise price | ||||||||||
Balance at January 1 |
| | € | |
| | € | |
| | € | | |||
Granted |
| | € | |
| | € | |
| | € | | |||
Forfeited |
| ( | € | |
| ( | € | |
| ( | € | | |||
Exercised |
| ( | € | |
| ( | € | |
| ( | € | | |||
Expired |
| ( | € | |
| ( | € | |
| ( | € | | |||
Balance at December 31 |
| | € | |
| | € | |
| | € | | |||
Exercisable at December 31 |
| |
|
|
| |
|
|
| |
|
| |||
The options outstanding at December 31, 2023 had an exercise price in the range of €
F-30
Movements in the number of RSUs outstanding are as follows:
| 2023 | 2022 | 2021 | |||
|
| Number of |
| Number of |
| Number of |
RSUs | RSUs | RSUs | ||||
Balance at January 1 |
| |
| |
| — |
Granted |
| |
| |
| |
Forfeited |
| ( |
| ( |
| ( |
Released |
| ( |
| ( |
| — |
Balance at December 31 |
| |
| |
| |
Refer to Note 27 for the share-based compensation granted to key management personnel.
14. Borrowings
|
| December 31, |
| December 31, |
2023 | 2022 | |||
| (€ in thousands) | |||
Innovation credit |
| | | |
Accrued interest on innovation credit | | | ||
Convertible loans | — | | ||
Accrued interest on convertible loans |
| — | | |
Total borrowings | | | ||
Current portion | — | ( | ||
Total non-current borrowings |
| |
| |
Innovation credit (“Innovatiekrediet”)
On December 10, 2018 ProQR was awarded an Innovation credit for the sepofarsen program. Amounts were drawn under this facility from 2018 through 2022. The credit of €
In December 2023, ProQR received a conditional waiver of the €
The amounts receivable relating to development & regulatory milestone payments under the Amended and Restated Asset Purchase Agreement with Laboratoires Théa S.A.S. (“Théa”) are subject to a right of pledge for the benefit of the Rijksdienst voor Ondernemend Nederland (“RVO”).
Convertible loans: Pontifax and Kreos
In July 2020, the Company entered into a convertible debt financing agreement with Pontifax Medison Debt Financing. Under the agreement, the Company had access to up to $
A second close of the convertible debt financing agreement was completed in August 2020 with Kreos Capital. Under the second agreement, the Company had access to up to €
F-31
In connection with the loan agreement, the Company issued to Pontifax and Kreos warrants to purchase up to an aggregate of
On December 29, 2021, the Company amended its convertible debt financing agreement with the Lenders. Under the amended agreement the Company drew down an additional $
In connection with the amended loan agreement, the Company issued to the Lenders warrants to purchase up to an aggregate of
The convertible loans from Pontifax and Kreos bore an interest of
In September 2022, ProQR extinguished its debt with Pontifax and Kreos by repaying all outstanding principal amounts. In addition, an early repayment penalty was incurred. The financial liability relating to Pontifax’ conversion options was derecognized from derivative financial instruments. The option premium on convertible loans relating to Kreos’ conversion options was derecognized from equity. The results related to the derecognition of these financial liabilities are disclosed in the table further below in this note.
Pontifax’ and Kreos’ warrants remain in place until their
Convertible loans: Amylon
Convertible loans amounting to €
In the third quarter of 2023, Amylon was legally dissolved. The effect of the resulting derecognition of Amylon’s remaining assets and liabilities is included in profit and loss as ‘result on derecognition of subsidiary’.
The results related to the derecognition of financial liabilities, as described above, are as follows:
|
| 2023 | 2022 | |
(€ in thousands) | ||||
Gain on waiver of Amylon convertible loans | | | ||
Loss on extinguishment of Pontifax and Kreos convertible loans | - | ( | ||
| ( | |||
F-32
Reconciliation of movements of liabilities to cash flows arising from financing activities
|
| Innovation |
| Convertible | Lease | |
credit | loans | liability | ||||
| (€ in thousands) | |||||
Balance at January 1, 2022 | | | | |||
Changes from financing cash flows | ||||||
Repayment of convertible loans | — | ( | — | |||
Repayment of lease liability | — | — | ( | |||
The effect of changes in foreign exchange rates | — | | — | |||
Other changes | ||||||
Interest expense | | | — | |||
Interest paid | — | ( | — | |||
Transaction costs | — | | — | |||
Repayments allocated to option premium on convertible loans (equity) | — | | — | |||
Repayments recognized as result on derecognition of financial liabilities | — | | — | |||
Effect of waived loan agreements | — | ( | — | |||
Effect of lease amendments | — | — | | |||
Balance at January 1, 2023 |
| | | | ||
Changes from financing cash flows | ||||||
Repayments | ( | — | ( | |||
The effect of changes in foreign exchange rates | — | — | — | |||
Other changes | ||||||
Interest expense | | | — | |||
Interest paid | — | — | — | |||
Effect of waived loan agreements | — | ( | — | |||
Effect of lease amendments | — | — | | |||
| ||||||
Balance at December 31, 2023 |
| |
| — | | |
F-33
15. Deferred Income
The following table summarizes details of deferred income at December 31, 2023 and December 31, 2022. The nature of the deferred income relating to Eli Lilly is described in Note 17.
|
| December 31, |
| December 31, |
2023 | 2022 | |||
| (€ in thousands) | |||
Eli Lilly up-front payment and premium on equity consideration |
| | | |
Total deferred income | | | ||
Current portion | ( | ( | ||
Total non-current deferred income |
| |
| |
The current portion of deferred income reflects the estimated value of the Company’s work under the Lilly collaboration that is expected to be performed within one year after the balance sheet date.
The table below analyzes ProQR’s undiscounted deferred income into relevant maturity groupings based on the remaining period at year-end until the contractual maturity date:
|
| Less than |
| Between 1 |
| Between 2 |
| |
1 year | and 2 years | and 5 years | Over 5 years | |||||
(€ in thousands) | ||||||||
At December 31, 2023 |
|
|
|
|
|
|
|
|
Deferred Income |
| | | | — | |||
Total |
| |
| |
| |
| — |
At December 31, 2022 |
|
|
|
|
|
|
|
|
Deferred Income |
| | | |
| — | ||
Total |
| |
| |
| |
| — |
16. Other Current Liabilities
At December 31, 2023, other current liabilities amount to €
F-34
17. Revenue
The following table summarizes details of revenue recognized in the years ended December 31, 2023, 2022 and 2021 by collaboration agreement and by category of revenue: upfront payments, other research and development service fees and equity consideration.
| 2023 |
| 2022 |
| 2021 | |
(€ in thousands) | ||||||
Up-front payments | ||||||
Eli Lilly |
| | | | ||
Yarrow |
| — | | | ||
Other R&D services | ||||||
Eli Lilly | — | | — | |||
Yarrow | — | | | |||
Equity consideration | ||||||
Eli Lilly | | | | |||
Yarrow | — | | | |||
| |
| |
| | |
The table below summarizes the changes in current and non-current deferred revenue for the years ended December 31, 2023, 2022 and 2021.
| Eli Lilly |
| Yarrow | |
(€ in thousands) | ||||
Balance at January 1, 2022 |
| | | |
Received or receivable | ||||
Upfront payment | | — | ||
Other R&D services | | | ||
Equity consideration | ( | — | ||
Revenue recognition | ||||
Upfront payment | ( | ( | ||
Other R&D services | ( | ( | ||
Equity consideration | ( | ( | ||
Foreign currency translation effects | ( | | ||
Balance at January 1, 2023 | | — | ||
Received or receivable | ||||
Upfront payment | — | — | ||
Equity consideration | — | — | ||
Revenue recognition | ||||
Upfront payment | ( | — | ||
Equity consideration | ( | — | ||
Foreign currency translation effects | | — | ||
Balance at December 31, 2023 |
| |
| — |
Eli Lilly collaboration
In September 2021, the Company entered into a global licensing and research collaboration with Lilly focused on the discovery, development, and commercialization of potential new medicines for genetic disorders in the liver and nervous system. ProQR and Lilly will use ProQR’s proprietary Axiomer RNA editing platform to progress new drug targets toward clinical development and commercialization.
F-35
Under the terms of the agreement, ProQR received an upfront payment and equity consideration, and is eligible to receive milestone payments and royalties on the net sales of any resulting products. In September 2021, the Company issued
With regard to its original collaboration with Lilly, the Company concluded as follows:
| ● | There is |
| ● | The transaction price of this agreement currently only includes fixed components, consisting of an up-front fee and an equity component. The agreement also contains variable components, but those are not yet included in the transaction price. Milestone payments will only be included to the extent that it is highly probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the milestones is subsequently resolved. Sales-based milestones and sales-based royalties will be included as the underlying sales occur. |
| ● | The Company recognizes revenue over time, using an input method that estimates the satisfaction of the performance obligation as the percentage of labor hours incurred compared to the total estimated labor hours required to complete the promised services. |
In December 2022, the Company and Lilly amended their research and collaboration agreement described above, which expanded the collaboration. Under the amended and restated research and collaboration agreement, Lilly will gain access to additional targets in the central nervous system and peripheral nervous system with ProQR’s Axiomer platform.
As described under Note 13, pursuant to the amended and restated agreement, the Company issued
With regard to the amended and restated research and collaboration agreement with Lilly, the Company concluded as follows:
| ● | There is |
| ● | The transaction price of this agreement currently only includes fixed components, consisting of an up-front fee and an equity component (discount). The agreement also contains variable components, but those are not yet included in the transaction price. Milestone payments will only be included to the extent that it is highly probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the milestones is subsequently resolved. Sales-based milestones and sales-based royalties will be included as the underlying sales occur. |
| ● | The Company recognizes revenue over time, using an input method that estimates the satisfaction of the performance obligation as the percentage of labor hours incurred compared to the total estimated labor hours required to complete the promised services. |
F-36
Yarrow Biotechnology collaboration
In May 2021, the Company entered into an exclusive worldwide license and discovery collaboration for an undisclosed target with Yarrow. Under the terms of the agreement, ProQR received an upfront payment, equity consideration and reimbursement for ongoing R&D services. ProQR was also eligible to receive milestone payments and royalties on the net sales of any resulting products. In May 2021, ProQR received an up-front payment of €
With regard to its collaboration with Yarrow, the Company concluded as follows:
| ● | There is |
| ● | The transaction price of this agreement currently includes both fixed and variable components. The fixed part consists of an up-front fee and an equity component. The variable part consists of a cost reimbursement for research and development activities. The agreement also contains other variable parts, but those are not yet included in the transaction price. Milestone payments will only be included to the extent that it is highly probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the milestones is subsequently resolved. Sales-based milestones and sales-based royalties will be included as the underlying sales occur. |
| ● | The Company recognizes revenue over time, using an input method that estimates the satisfaction of the performance obligation as the percentage of labor hours incurred compared to the total estimated labor hours required to complete the promised services. |
The Yarrow collaboration was terminated in the third quarter of 2022.
18. Other Income
| 2023 |
| 2022 |
| 2021 | |
(€ in thousands) | ||||||
Net gain on divestment of intellectual property | | — | — | |||
Grant income |
| |
| |
| |
Other income | | | | |||
| |
| |
| | |
In 2023, ProQR completed the divestment of its late-stage ophthalmic intellectual property assets, sepofarsen and ultevursen, to Théa. Under the terms of the agreement, ProQR received an initial payment of €
On February 9, 2018, the Company entered into a partnership agreement with FFB, under which FFB agreed to provide funding of $
F-37
19. Operating Costs
Total operating costs include the following expenses by nature:
| 2023 |
| 2022 |
| 2021 | |
(€ in thousands) | ||||||
Employee benefits |
| | | | ||
External R&D costs | | | | |||
Laboratory costs and other consumables | | | | |||
Advisory and legal costs | | | | |||
Insurance costs | | | | |||
Depreciation | | | | |||
Patent and license expenses | | | | |||
Other | | | | |||
| |
| |
| | |
Reclassifications
Certain prior year amounts have been reclassified to conform to current year presentation.
20. Employee Benefits
| 2023 |
| 2022 |
| 2021 | |
(€ in thousands) | ||||||
Wages and salaries |
| |
| |
| |
Social security costs |
| |
| |
| |
Pension costs — defined contribution plans |
| |
| |
| |
Equity-settled share based payments |
| |
| |
| |
| |
| |
| | |
Average number of employees for the period |
| |
| |
| |
Employees per activity at December 31 (converted to FTE):
|
| December 31, 2023 |
| December 31, 2022 |
| December 31, 2021 |
Research and Development |
| |
| |
| |
General and Administrative |
| |
| |
| |
Total number of employees (converted to FTE) |
| |
| |
| |
Of all employees
Included in the wages and salaries for 2023 is a credit of €
F-38
21. Financial Income and Financial Expense
| 2023 |
| 2022 |
| 2021 | |
(€ in thousands) | ||||||
Interest income: |
|
|
|
|
|
|
Current accounts and deposits |
| |
| |
| |
Interest costs: |
|
|
|
|
|
|
Current accounts and deposits | ( | ( | ( | |||
Lease liability | ( | ( | ( | |||
Loans and borrowings |
| ( |
| ( |
| ( |
Foreign exchange result: |
|
|
|
|
|
|
Net foreign exchange benefit/(loss) |
| ( |
| |
| |
| |
| ( |
| ( | |
Financial income amounting to €
22. Results related to financial liabilities measured at fair value through profit or loss
Results related to financial liabilities measured at fair value through profit or loss represent changes in the fair value of derivative financial instruments since their initial recognition. These derivative financial instruments consist of conversion options and warrants issued in connection with our convertible loans, which are described in Note 14.
At
23. Income Taxes
The calculation of the tax charge is as follows:
| 2023 |
| 2022 |
| 2021 |
| |
(€ in thousands) |
| ||||||
Consolidated result before corporate income taxes | ( | ( | ( | ||||
Exclude: results related to associates | — | ( | ( | ||||
( | ( | ( | |||||
Income tax based on domestic rate (2023 and 2022: |
| |
| |
| | |
Tax effect of: |
|
|
| ||||
Different tax rates in foreign jurisdictions | ( | | | ||||
Non-deductible expenses / non-taxable gains |
| ( |
| |
| ( | |
Share- and loan-issue expenditures that are taxable | — | — | | ||||
Change in unrecognized deductible temporary differences |
| ( |
| ( |
| ( | |
Current year losses for which no deferred tax asset was recognized |
| ( |
| ( |
| ( | |
True-up for prior year |
| |
| ( |
| ( | |
Income tax benefit / (charge) |
| |
| ( |
| ( | |
Effective tax rate |
| — | % | — | % | — | % |
F-39
The Company recognizes deferred tax assets arising from unused tax losses, deductible temporary differences or tax credits only to the extent that the Company has sufficient taxable temporary differences or there is convincing evidence that sufficient taxable profit will be available against which the unused tax losses or unused tax credits can be utilized. Management’s judgment is that such convincing evidence is currently not sufficiently available and a deferred tax asset is therefore only recognized to the extent that the Company has sufficient taxable temporary differences. Consequently, the Company has not recognized a deferred tax asset related to operating losses.
A deferred tax liability amounting to €
As per December 31, 2023, the Company has a total amount of €
24. Earnings Per Share
(a) Basic and diluted earnings per share
Basic earnings per share are calculated by dividing the result attributable to owners of the Company by the weighted average number of shares outstanding during the year.
|
| 2023 |
| 2022 |
| 2021 | |||
Result attributable to owners of the Company (€ in thousands) |
| ( |
| ( |
| ( | |||
Weighted average number of shares outstanding |
| |
| |
| | |||
Basic (and diluted) earnings per share (€ per share) | € | ( | € | ( | € | ( | |||
(b) Diluted earnings per share
For the periods included in these financial statements, the share options are not included in the diluted earnings per share calculation as the Company was loss-making in all periods. Due to the anti-dilutive nature of the outstanding options, basic and diluted earnings per share are equal.
(c) Dividends per share
The Company did not declare dividends for any of the years presented in these financial statements.
25. Leases
The Company leases office and laboratory facilities of
F-40
The initial
The initially recognized lease liability and the corresponding right-of-use asset for this lease contract, on July 1, 2020, amounted to €
Annually in June, the lease price is amended to reflect an indexation. In June 2023, the lease liability was remeasured, resulting in an increase in the carrying amounts of the lease liability and the right-of-use of €
The following table summarizes the relevant disclosures in relation to our leases in 2023, 2022 and 2021:
| 2023 | 2022 | 2021 | |||
(€ in thousands) | (€ in thousands) | (€ in thousands) | ||||
Depreciation charge for right-of-use assets |
| | | | ||
Interest expense on lease liabilities |
| | | | ||
Expense relating to short-term leases |
| | | | ||
Total cash outflow for leases |
| | | | ||
Additions to right-of-use assets during the period | | | |
The carrying amount of the right-of-use asset at the end of the reporting period is disclosed in Note 7 Property, Plant & Equipment.
A maturity analysis of our lease liability is included in Note 5 Financial Risk Management under (c) Liquidity risk. The total undiscounted commitment for lease agreements to which the Company had committed at December 31, 2023 amounts to €
26. Commitments and Contingencies
(a) Claims
There are
(b) Patent license agreements
On October 29, 2018, ProQR signed an agreement with Ionis Pharmaceuticals (“Ionis”) to license QR-1123 (formerly “IONIS-RHO-2.5Rx”), an RNA medicine for autosomal dominant retinitis pigmentosa (“adRP”) caused by the P23H mutation in the rhodopsin (“RHO”) gene. Under the terms of the agreement, ProQR was granted an exclusive worldwide license to QR-1123 and relevant patents. In 2018, ProQR paid the first installment of an upfront payment in ordinary shares in the aggregate amount of $
F-41
In April 2014, the Company entered into a Patent License Agreement with Radboud University Medical Center (“Radboud”) in the field of antisense oligonucleotide-based therapy for Leber congenital amaurosis (“LCA”). Under the terms of this license agreement, the Company has an exclusive, sublicensable, world-wide royalty-bearing license under certain Radboud patent rights to develop, make, have made, use, sell, offer for sale and import certain licensed products of Radboud for use in all prophylactic and therapeutic uses in the field of LCA. This license is assigned in full per December 2023 as part of the divestment of the product sepofarsen.
In June 2015, the Company entered into another license agreement with Radboud. Under the terms of this license agreement, the Company has an exclusive, sublicensable, world-wide royalty-bearing license under certain Radboud patent rights to develop, make, have made, use, sell, offer for sale and import certain licensed products of Radboud for use in all prophylactic and therapeutic uses in the field of Usher syndrome. This license was assigned in full per December 2023 as part of the divestment of the product ultevursen.
In January 2018, the Company entered into a license agreement with Inserm Transfert SA and Assistance-Publique-Hôpiteaux de Paris. Under the terms of the agreement, the Company has a world-wide, exclusive, royalty-bearing license under patent rights belonging to Inserm Transfert SA and other co-owners to develop, have developed, make, have made, use, have used and sell, have sold or otherwise distribute certain licensed products related to antisense oligonucleotides for treating LCA and method of treatment claims relating to modulation of the splicing of the CEP290 gene product. This license agreement is assigned per December 2023 in connection with the sale of the ophthalmology products, sepofarsen and ultevursen. In consideration for the assignment, the Company has agreed to accept certain royalty obligations upon sepofarsen reaching certain regulatory milestones and net sales of products sold.
In January 2017, the Company entered into an agreement with the Leiden University Medical Center (“LUMC”), which gives us a world-wide, exclusive, royalty-bearing license in the field of Huntington’s disease, under certain patent rights of LUMC regarding antisense oligonucleotide based therapies. This license agreement contains certain diligence obligations for the Company coupled to milestone payments and complements the Company’s intellectual property relating to the HD program. This license was terminated per July 2023.
In February 2019, the Company entered into an agreement with the University of Rochester, New York, which gives us a world-wide, exclusive, royalty-bearing, sublicensable license in the field of antisense oligonucleotides for use in nucleotide specific RNA editing through pseudouridylation, under certain patent rights of University of Rochester. This license agreement contains certain diligence obligations for the Company coupled to milestone payments and complements the Company’s intellectual property relating to the Axiomer/pseudouridylation program.
In September 2020, the Company entered into an agreement with Vico Therapeutics B.V., which gives us a world-wide, exclusive, royalty-bearing, sublicensable license in the field of the prophylactic and therapeutic use of antisense oligonucleotide for the treatment of Fuch’s Endothelial Corneal Dystrophy caused by a trinucleotide repeat, under certain patent rights of Vico Therapeutics B.V. In partial consideration of the rights and licenses granted by the license agreement, the Company is required to make annual maintenance payments. Unless terminated earlier in accordance with the terms of the license agreement, the agreement will stay in effect until the expiration of all of the licensed patent rights. The license agreement may be terminated by either party in the event of an uncured breach by the breaching party. Vico Therapeutics B.V. may terminate the license agreement if the Company applies for an order or an order is made declaring the Company bankrupt or granting the Company suspension of payments, or a liquidator is appointed for the Company, or the Company is dissolved, liquidated, or ceases to carry on all or a substantial part of its business or a decision is taken to that effect, or in the event uncured payment defaults. The development of this candidate has been suspended per the strategic shift in focus as announced in August 2022.
(c) Clinical support agreements
On February 9, 2018, the Company entered into an agreement with FFB, under which FFB has provided funding of $
F-42
Pursuant to the terms of the agreement, we were obligated to make certain repayments to FFB subject to development milestones. In December 2023, upon the occurrence of the sale of ultevursen to Théa, these payables were settled by means of a lump-sum payment in the amount of €
(d) Research and development commitments
The Company has research and development commitments, mainly with CRO’s, amounting to €
27. Related-Party Transactions
Details of transactions between the Company and related parties are disclosed below.
(a) Compensation of the Supervisory Board
The remuneration of the supervisory board members in 2023 is set out in the table below:
2023 | ||||||||
| Short term |
| Post-employment |
| Share-based |
| ||
employee benefits | benefits | payment | Total | |||||
(€ in thousands) | ||||||||
Mr. Dinko Valerio |
| | — | | | |||
Mr. Antoine Papiernik* |
| — | — | — | — | |||
Ms. Alison F. Lawton |
| | — | | | |||
Mr. James Shannon |
| | — | | | |||
Mr. Bart Filius | | — | | | ||||
Ms. Begoña Carreño** | | — | | | ||||
Ms. Theresa Heggie*** | | — | | | ||||
| |
| — |
| |
| | |
* Mr. Papiernik stepped down from the supervisory board on May 18, 2023.
** Ms. Carreño was elected to the supervisory board on May 18, 2023. The remuneration set forth for Ms. Carreño in the table above covers the period from May 18, 2023 to December 31, 2023.
*** Ms. Heggie was elected to the supervisory board on May 18, 2023. The remuneration set forth for Ms. Heggie in the table above covers the period from May 18, 2023 to December 31, 2023. Ms. Heggie’s share-based payments include the effects of options and RSUs that were granted to her before her reappointment to the supervisory board on May 18, 2023.
F-43
The remuneration of the supervisory board members in 2022 is set out in the table below:
2022 | ||||||||
| Short term |
| Post employment |
| Share-based |
| ||
employee benefits | benefits | payment | Total | |||||
(€ in thousands) | ||||||||
Mr. Dinko Valerio |
| | — | | | |||
Mr. Antoine Papiernik |
| — | — | — | — | |||
Ms. Alison F. Lawton |
| | — | | | |||
Mr. James Shannon |
| | — | | | |||
Mr. Bart Filius | | — | | | ||||
| |
| — |
| |
| | |
The 2021 remuneration is set out in the table below:
2021 | ||||||||
| Short term |
| Post employment |
| Share-based |
| ||
employee benefits | benefits | payment | Total | |||||
(€ in thousands) | ||||||||
Mr. Dinko Valerio |
| | — | | | |||
Mr. Antoine Papiernik |
| — | — | — | — | |||
Ms. Alison F. Lawton |
| | — | | | |||
Mr. James Shannon |
| | — | | | |||
Mr. Bart Filius | | — | |
| | |||
Ms. Theresa Heggie* |
| | — | | | |||
|
| — |
| |
| | ||
* Ms. Heggie stepped down from the supervisory board on October 1, 2021, in connection with her appointment as Chief Commercial Officer of the Company. The remuneration set forth for Ms. Heggie in the table above covers the period from January 1, 2021 to October 1, 2021.
In 2023, 2022 and 2021, Mr. Papiernik waived his compensation.
As at December 31, 2023:
| ● | Mr. Dinko Valerio holds |
| ● | Mr. Antoine Papiernik does not hold any shares or options in the Company. As a managing partner of Sofinnova Partners SAS, the management company of Sofinnova Capital VII FCPR, holder of |
F-44
| ● | Ms. Alison F. Lawton holds |
| ● | Mr. James Shannon holds |
| ● | Mr. Bart Filius holds |
| ● | Ms. Begoña Carreño holds |
| ● | Ms. Theresa Heggie holds |
F-45
(b) Compensation of key management
Our management board is supported by our officers, or senior management. Mr. Daniel de Boer and Mr. Rene Beukema are the statutory directors of the Company. The total remuneration of the management board and senior management in 2023 amounted to €
2023 | ||||||||
| Short term |
| Post |
|
| |||
employee | employment | Share-based | ||||||
benefits | benefits | payment | Total | |||||
(€ in thousands) | ||||||||
Mr. D.A. de Boer1 |
| | | |
| | ||
Mr. R.K. Beukema1 | | | | | ||||
Management Board |
| |
| |
| |
| |
Senior Management |
| | | |
| | ||
| |
| |
| |
| | |
| 1 | Short term employee benefits include bonuses for Mr. Daniel de Boer of € |
The total remuneration of the management board and senior management in 2022 amounted to €
2022 | ||||||||
| Short term |
| Post |
|
| |||
employee | employment | Share-based | ||||||
benefits | benefits | payment | Total | |||||
(€ in thousands) | ||||||||
Mr. D.A. de Boer1 |
| | | |
| | ||
Mr. R.K. Beukema1 |
| | | |
| | ||
Management Board |
| |
| |
| |
| |
Senior Management |
| | | |
| | ||
| |
| |
| |
| | |
| 1 | Short term employee benefits include a bonus for Mr. Daniel de Boer of € |
The total remuneration of the management board and senior management in 2021 amounted to €
2021 | ||||||||
| Short term |
| Post |
|
| |||
employee | employment | Share-based | ||||||
benefits | benefits | payment | Total | |||||
(€ in thousands) | ||||||||
Mr. D.A. de Boer1 |
| | | |
| | ||
Management Board |
| |
| |
| |
| |
Senior Management |
| | | |
| | ||
| |
| |
| |
| | |
| 1 | Short term employee benefits include a bonus for Mr. Daniel de Boer of € |
F-46
As at December 31, 2023:
| ● | Mr. Daniel de Boer holds |
| ● | Mr. Rene Beukema holds |
ProQR does not grant any loans, advance payments and guarantees to members of the Management and Supervisory Board.
(c) Transactions with Yarrow Biotechnology, Inc.
As described in Note 8. Investments in Associates, the Company, as of October 2023, no longer has significant influence over Yarrow Biotechnology, Inc. Yarrow is therefore, no longer considered a related party as of that point onwards. The Company did not have any transactions with Yarrow in the year ended December 31, 2023. Transactions with Yarrow for the years ended December 31, 2022 and 2021 are described in Note 17. Revenue.
28. Auditor fees
The fees for services provided by our external auditors, KPMG Accountants N.V. for the years ended December 31, 2023, 2022 and 2021, are specified below for each of the financial years indicated:
| 2023 |
| 2022 |
| 2021 | |
(€ in thousands) | ||||||
Audit fees |
| |
| |
| |
Audit-related fees |
| — |
| |
| |
| |
| |
| | |
Audit fees consist of aggregate fees for professional services provided in connection with the annual audit of our financial statements. Audit-related fees consist of procedures relating to share offerings, such as comfort letters, as well as consents and review of documents filed with the SEC.
29. Subsequent events
No significant events occurred after the balance sheet date.
F-47